Mechanisms of Cardiac Arrhythmias
Augustus O. Grant
Sarfraz Durrani
Overview
Our understanding of the mechanisms of cardiac arrhythmias has advanced with the introduction of new technologies to study the electrical activity of the heart. These studies show that most arrhythmias are generated by the same ion channels that sustain normal activity. However, alterations in ion channel number and the intracellular and extracellular concentrations of ions in disease states provide the setting for arrhythmias. Structure is also very important, from the changes in protein structure in the long QT and Brugada syndromes to the remolding of the atria and ventricle in ischemia, cardiac hypertrophy and other forms of heart muscle disease. Reentry is the likely mechanism of most sustained arrhythmias in chronic heart disease. Abnormal impulse formation, as evidenced by early and delayed afterdepolarization, is the second major cause of arrhythmias. In some circumstances, such as focal atrial fibrillation, the arrhythmia may be initiated by abnormal impulse formation and sustained by reentry. The clinical setting—family history, drug use, electrolyte imbalance, and electrocardiographic (ECG) findings—may suggest a mechanism. Antiarrhythmic drugs act on membrane ion channels. Specificity, selectivity, and therapeutic index are low. As a result, physical strategies such as ablation and cardioverter defibrillator implantation are playing an increasingly important role in arrhythmia management.
Historical Perspective
This chapter reviews normal impulse generation and propagation in the heart and the ways that disturbances of either mechanism result in cardiac arrhythmias. Emphasis is placed on observations made at the cellular and subcellular levels. Analysis of the mechanisms of arrhythmias at the higher levels of tissues and in the in situ heart is the focus of Chapters 60 and 62.
The earliest direct recording of electrical activity of the heart used sensitive string galvanometers to record unipolar and bipolar electrogram to study regional spread of excitation. Lewis and colleagues used accurate extracellular recordings to relate propagation velocity to structure. The millimeter resolution of such recording has been replaced by voltage-sensitive dyes with a resolution of microns. These strategies have permitted the mapping of excitation spread in the basal state and during arrhythmias. The basis for normal and abnormal excitation had to await techniques that permitted transmembrane potential recordings. Ling-Gerrard capillary microelectrodes permitted transmembrane action potential recordings from groups of cells with little apparent damage to the cells. Those studies documented the relationship between the initial QRS complex and the T wave on the ECG to periods of rapid depolarization (phase 0) and repolarization (phase 3) of the action potential. Analysis of the ionic basis of the action potential proved more formidable. Membrane potential (V) was varying with time (dV/dT greater than 0) as excitation propagated over the surface of the heart (dV/dX greater than 0). Marmont and Cole recognized that the analysis of excitation required that one of the independent variables (time t, or distance x) had to be controlled (zero) as the experimenter varied V (2,3). These investigators developed the voltage clamp technique, which permitted measurement of the underlying current as a function of time as membrane voltage V was varied. This required that V should not vary with distance (i.e., dV/dX = 0, space clamping). The complexity of naturally occurring cardiac preparation
severely limited space clamping. However, the preparation of single calcium-tolerant cardiac myocytes by Powell and Twist enabled a quantitative voltage clamp of the major currents that contribute to the action potential (4).
severely limited space clamping. However, the preparation of single calcium-tolerant cardiac myocytes by Powell and Twist enabled a quantitative voltage clamp of the major currents that contribute to the action potential (4).
Neher and Sakmann introduced the patch clamp technique that permitted quantitative voltage clamp of even the smallest cardiac cells such as those of the sinoatrial (SA) and atrioventricular (AV) nodes (5). The technique also permitted the resolution of current through single ion channels. All the major ion channels that contribute to the generation of the action potential have now been cloned and functionally expressed. Subgroups of arrhythmias occurring in the structurally normal heart have been shown to be monogenetic, arising from mutations of ion channel genes (6,7). The more common arrhythmias, such as ischemic-induced ventricular tachycardia and atrial fibrillation, are of multifactorial origin, with susceptibility genes playing an important role. Analysis of these and other arrhythmias starts with the premise that aberration of the normal mechanisms of impulse initiation and propagation are central to the genesis of arrhythmias. Therefore, normal impulse initiation and propagation are the starting points for the analysis of arrhythmia mechanisms.
Basic Electrophysiology of the Action Potential
The five phases—0, 1, 2, 3, and 4 — of the action potential recorded from a ventricular cell are illustrated in Figure 57.1. Phase 4 describes the membrane potential during diastole. Working myocardial cells maintain a steady diastolic level of approximately –85 mV. Conduction system myocytes in the SA and AV nodes, excitable cells of the mitral and tricuspid valve, the pulmonary veins, and the coronary sinus exhibit slow diastolic depolarization during phase 4. Excitation of resting cells results in a rapid transition from phase 4 to phase 0. This phase represents a period of rapid depolarization resulting in reversal of the transmembrane potential, and lasts less than 1 millisecond. Phase 1 is a period of rapid repolarization that returns the membrane to the level of the plateau. This phase (1) shows the greatest variability among various regions of the heart and among various species. It is prominent in Purkinje and epicardial cells, but it is diminutive or absent in endocardial cells. Phase 2 is the plateau phase of the action potential and lasts up to several hundred milliseconds. It is the phase that most clearly distinguishes the cardiac action potential from the brief action potentials of skeletal muscle and nerve. Phase 3 marks the period of terminal repolarization when the membrane potential is restored to its resting level.
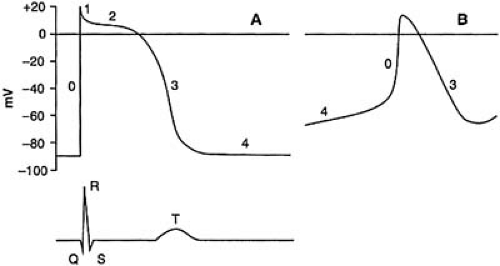 FIGURE 57.1. Idealized action potentials of nonpacemaker and pacemaker cells. A: The action potential of a nonpacemaker cell is shown. Five phases of the action potential, 0, 1, 2, 3, and 4 can be identified. The membrane potential during phase 4 is stable at approximately –90 mV. The electrocardiogram in the lower section of A shows the relationship to the various phases of the action potential: The QRS complex corresponds to phase 0 and the T wave to phase 3. The QT interval approximates the action potential duration. B: The action potential from a pacemaker cell is shown. The membrane potential gradually declines during phase 4. |
TABLE 57.1 Intracellular and Extracellular Ion Concentrations in Cardiac Muscle | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
An uneven (nonequilibrium) distribution of ions across the cell membrane is an essential basis for the action potential and is summarized in Table 57.1. Under physiologic conditions, the sodium/potassium (Na+/K+) pump moves three Na+ ions out of the cell in exchange for two extracellular K+ ions; one molecule of adenosine triphosphate (ATP) is consumed in the process. Because the stoichiometry of ion movement is not 1:1, the Na+/K+ pump generates outward current. The overdrive suppression of pacemaker activity following rapid stimulation results from the hyperpolarizing effect of the Na+/K+ pump current. Arrhythmias resulting from digitalis intoxication are secondary consequences of pump inhibition. The Na+/calcium (Na+/Ca2+) exchanger uses the Na+ gradient established by the Na+/K+ pump to transport Ca+ across the cell membrane. The Na+/Ca2+ exchanger transports Ca2+ into the cell at plateau levels of membrane potential and Ca2+ out of the cell during diastole. Because the stoichiometry of the exchanger is approximately 3:1, it is also electrogenic. The Na+/H+ and bicarbonate/chloride (HCO3–/Cl–) exchangers also contribute to the uneven distribution of ions across the cell membrane.
During the action potential, selectively permeable ion channels open or close, and the direction of ion movement (and the resulting current) is determined by the ratio of the intracellular and extracellular concentration of each ion and the membrane potential. Ion movement is passive. We now review the ion channels that are crucial to each phase of the action potential in more detail.
Phase 1 of the Action Potential
Nonpacemaker Cells
For cells with a stable resting (phase 4) membrane potential, the inward rectifier K+ channel IK1 has the highest conductance. The membrane potential Em is only a few millivolts positive to the equilibrium potential for K+, EK. The small deviation between Em and EK reflects the limited permeability of the IK1 channel to Na+ and the finite conductance of other ion channels at the level of the resting potential. Elevation of the extracellular K reduces the membrane potential according to the Nernst relationship:
where R is the universal gas constant, T the absolute temperature, F the Faradays constant, and [Ko+] and [K1+] the
extracellular and intracellular K+ concentrations, respectively. The conductance of most ion channels is independent of the electrochemical driving force (Em - Ex) acting on the permeant ion x. In the case of the inward rectifier, channel conductance is increased by elevations in extracellular K+ concentration. Marked elevation in [Ko+] around cells is an early event during myocardial ischemia and may play a critical role in arrhythmias associated with acute myocardial ischemia.
extracellular and intracellular K+ concentrations, respectively. The conductance of most ion channels is independent of the electrochemical driving force (Em - Ex) acting on the permeant ion x. In the case of the inward rectifier, channel conductance is increased by elevations in extracellular K+ concentration. Marked elevation in [Ko+] around cells is an early event during myocardial ischemia and may play a critical role in arrhythmias associated with acute myocardial ischemia.
Pacemaker Cells
Cells in the SA and AV nodes and the His-Purkinje system undergo slow diastolic depolarization during phase 4. Three ionic currents contribute to this depolarization: (a) a background inward current carried by Na+ ions; (b) the delayed rectifier current activated during the preceding depolarization; and (c) a hyperpolarization-activated current, If, carried by Na+ and K+ ions. Pacemaker activity in the SA and AV nodes is primarily dependent on a and b, whereas c plays a more important role in Purkinje’s cells (8,9). The relative conductance of other ion channels compared with that of the inward rectifier K+ channel is substantial in pacemaker cells. These cells are much less sensitive to elevation of [Ko+], and this characteristic may explain the persistence of SA and AV node function in states of marked hyperkalemia.
The rate of discharge of pacemaker cells may be slowed by increasing the maximum diastolic potential, decreasing the slope of phase 4, or decreasing the threshold potential; the opposite changes accelerate discharge rate. Sympathetic stimulation and parasympathetic stimulation change pacemaker rates by two mechanisms: (a) they shift the primary site of the pacemaker within the SA node and (b) they modify the kinetics of the currents that control pacemaker activity. Vagal stimulation produces a caudal shift in the site of the primary pacemaker; sympathetic stimulation produces a cephalad shift. Acetylcholine increases the maximum diastolic potential by activating the inward K+ current IKACh. Acetylcholine also shifts the voltage dependence of If activation to more negative potentials. As a result, a smaller component of If is activated during the pacemaker potential. β-Adrenergic stimulation has the opposite effect on the activation of If.
Phase 0 of the Action Potential
Nonpacemaker Cells
In nonpacemaker cells, an inward voltage-dependent Na current is responsible for phase 0 of the action potential. It is the prototype of voltage-dependent ion channels. The properties of the Na+ channel are described in detail to illustrate some of the general properties of voltage-dependent ion channels. Two distinct properties, gating and ion permeation, define the behavior of ion channels. Gating describes the opening and closing of the ion channel. Voltage-gated channels such as the Na+ channel have an intrinsic charge and respond to membrane potential with a change in conformational state. During phase 4, Na+ channels are closed. They open transiently (<1 millisecond) in response to membrane depolarization. The generally accepted gating model is based on the seminal work of Hodgkin and Huxley (10) in the 1950s and is illustrated in Figure 57.2. Ion movement through the Na+ channel is under the control of two gates: the activation, or m, gates and the inactivation, or h, gate. At normal resting potentials, the activation gates are closed and the inactivation gate is open. The activation gates open during depolarization, and Na+ ions move into the cell down their electrochemical gradient. Closure of the inactivation gate follows with a brief delay, and ion permeation is terminated. Repolarization to high levels of membrane potential is required for recovery from inactivation.
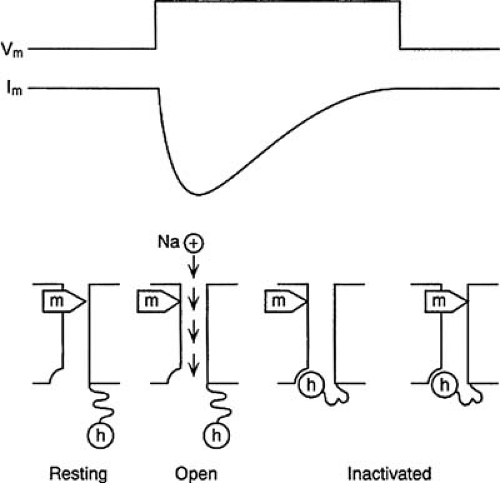 FIGURE 57.2. The Hodgkin-Huxley model of sodium ion (Na+) channel gating. The upper trace (Vm) shows a voltage step, and the middle trace (Im) shows the resulting membrane current. A physical model to account for the transient current is shown in the lower trace. The Na+ channel is represented as a pore spanning the membrane. In the resting state, the activation (m) gate is in the closed position, and the inactivation (h) gate is in the open position. Following depolarization, the m gate assumes the open position. With both gates open, Na+ ions move into the cell. The h gate then moves into the closed position, blocking ion movement. When the membrane is returned to the resting level, the m gate moves into the closed position, a process termed deactivation. After a variable interval, the h gate moves into the open position, a process termed recovery from inactivation (not shown). |
Membrane depolarization decreases the fraction of Na+ channel available for opening; at a membrane potential of approximately –75 mV, only 50% of Na+ channels are available for opening. This voltage-dependent inactivation is important in the genesis of arrhythmias and in antiarrhythmic drug action. Most membrane depressants, such as hyperkalemia and acidosis associated with ischemia and myocardial stretch, cause membrane depolarization and Na channel inactivation. Class I antiarrhythmic drugs decrease the availability of Na+ channels at all membrane potentials. Some drugs such as lidocaine also bind preferentially to inactivated Na+ channels and are therefore more effective in blocking conduction in partially depolarized ischemic tissue.
Ion permeation is the second important property of ion channels. The selective permeability to specific ions forms the most common basis for the classification of ion channels. The Na+ channel has a selectivity ratio for Na+ to K+ of 12:1. The simplest ion channel would permit inward and outward ion movement with equal facility. This is not the case with many ion channels. A channel is said to rectify in the inward direction if a voltage displacement produces a larger current when ions are moving in the inward direction compared with the outward current produced by a voltage displacement of the same amplitude but opposite direction. Inward rectification is a common property of K+ channels. Depolarization to positive potentials should markedly increase outward K+ current as the membrane potential moves positive to the K+ equilibrium potential. However, K+ current decreases at depolarized potentials because of inward rectification. Inward rectification of some K+ channels results from blockade by intracellular polyamines and magnesium (Mg2+) (11).
Pacemaker Cells
At the depolarized level of the maximum diastolic potential of pacemaker cells (approximately –60 mV), the Na+ channels are inactivated. Inward Ca2+ current is responsible for phase 0 of the action potential. Of the two types of Ca2+ channels in cardiac muscle, L and T, it is the former that makes the major contribution to phase 0. Like Na+ channels, Ca2+ channels exist in resting, open, and inactivated states. However, the magnitude, kinetics, and ion permeation characteristics differ from the Na+ channel: (a) peak amplitude is less than 10% that of the Na+ current, (b) activation and inactivation occur at more depolarized potentials and are somewhat slower, and (c) Ca2+ is the only physiologic charge carrier.
Phase 2 of the Action Potential
A transient, outward K+ current is responsible for phase 1 of the action potential. Calcium-dependent and calcium-independent K+ channels are responsible for this current component. The distribution of these channels is consistent with the magnitude of phase 1 of the action potential: the channel density is high in the epicardium and is low in the endocardium. A reduction in the magnitude of the transient outward current is consistently observed in left ventricular hypertrophy and congestive heart failure (12). The resulting prolongation of the action potential duration (APD) may account for the increased susceptibility to arrhythmias in these states.
Phases 2 and 3 of the Action Potential
Phase 2 of the action potential may last tens (atrium) to hundreds of milliseconds (His-Purkinje system and ventricle). A delicate balance of inward and outward currents results in little change in membrane potential. The principal inward currents are the L-type Ca2+ current and a small residual component of inward Na+ current. The significance of the latter has been highlighted by studies of the molecular basis of the long QT syndrome. Several kindreds have been identified in whom the genetic defect has been pinpointed to the locus on chromosome 3 that encodes the cardiac Na+ channel (13). Genetic defects that have been identified include single amino acid substitutions and a three–amino acid deletion (13,14). The consequence of the defect is a failure of or reversibility of inactivation resulting in an increase in the magnitude of the residual slow component of Na+ current during phase 2. The increased inward current prolongs the duration of action potential and the QT interval on the surface ECG.
A delayed rectifier current, consisting of rapid and slow components IKr and IKs, is activated at depolarized potentials. IKr is activated rapidly, shows inward rectification, and is unchanged by β-adrenergic stimulation. IKs is approximately 10 times larger than IKr, does not rectify, and is increased by β-adrenergic stimulation. Mutations in the genes encoding IKr and IKs or their regulatory subunits may result in a reduction of outward K current, APD and QT interval prolongation, and torsades de pointes. The Na+/Ca2+ exchanger and the Na+/K+ pump provide minor current components during phase 2. The activation of IKr and IKs initiates phase 3 repolarization. As the resting potential is approached, IK1 makes an increasingly important contribution to the net membrane current and hence to terminal repolarization.
Other Ion Channels Contributing to Multiple Phases of the Action Potential
Certain ion channels make a contribution to the action potential only under special circumstances. KATP channels are present in high density in cardiac cells. Opening of the KATP channels depends on the ratio of adenosine diphosphate (ADP) to ATP. Micromolar concentrations of ATP close the channels. Reduction in the ATP concentration during ischemia causes a small fraction of these channels to open. Because of their high density, substantial outward current is produced, markedly reducing the APD. As K+ ions escape into a restricted extracellular space, [K+]o rises substantially. Sulfonylureas open these channels and shorten the APD. Cl–-selective channels are present in cardiac cells. Under normal circumstances, they do not account for significant membrane current. However, the Cl– current is markedly activated by sympathetic stimulation and may play a role in repolarization under these circumstances.
Excitability
Excitability is the ability of a cell or group of cells to respond to a stimulus with a regenerative action potential. The membrane potential must be displaced from its resting level, Vr, to a threshold potential, VTH, at which the inward current (Na+ or Ca2+) becomes regenerative. Excitability depends on the passive and active properties of the cell membrane. The passive properties include the membrane resistance and capacitance and the intercellular resistance. A certain minimum charge has to be applied to the cell membrane to elicit a regenerative response. Excitability is inversely related to the charge required for excitation. The relationship between stimulus amplitude and duration is summarized in the familiar strength-duration curve shown in Figure 57.3. As stimulus duration is increased, the threshold stimulus declines and approaches an asymptotic value at long times. The voltage-dependent properties of the inward current determine the threshold potential.
Extracellular electrolytes such as K+ and antiarrhythmic drugs have a complex effect on excitability. Domiguez and Fozzard (15) showed that small elevations of [K+]o increase excitability by reducing the difference between the resting and threshold potentials (Vr – VTH); large elevations of [K+]o reduce excitability by inducing Na+ channel inactivation. Antiarrhythmic drugs influence excitability by changing both the passive membrane properties and the inward Na+ current. Na+ channel blockers reduce excitability, shifting the curve upward and to the right. K+ channel blockers shift the curve in the opposite direction and increase excitability. These changes in
excitability are reflected in the energy requirements for internal and external defibrillation.
excitability are reflected in the energy requirements for internal and external defibrillation.
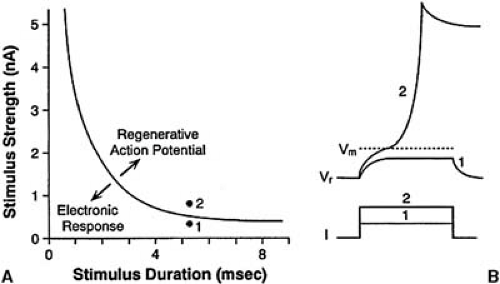 FIGURE 57.3. Stimulus strength-duration relationship of a ventricular cell. A: The strength-duration curve represents the minimum stimulus strength (ordinate) required to elicit a regenerative action potential as a function of stimulus duration (abscissa). B: Effect that a subthreshold stimulus (point 1) and suprathreshold stimulus (point 2) elicit on the membrane potential. The subthreshold stimulus elicits a nonregenerative (electronic) membrane depolarization. The suprathreshold stimulus elicits a regenerative action potential. |
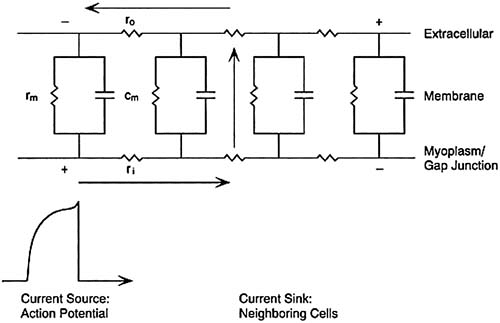 FIGURE 57.4. Equivalent circuit model of a cardiac fiber. The components of cardiac cells are modeled as equivalent electrical circuit elements. The unit membrane resistance is rm. The intracellular resistance (ri) consists of the series resistance of the myoplasm and gap junctions. The resistance of the extracellular space is ro. cm is the unit membrane capacitance. The action potential on the far left is the source of current for excitation of this patch of membrane. The current pathway is through ri, rm, cm, and ro, completing the circuit back to the source. |
Excitability is particularly important in the analysis of the ability of cardiac cells to undergo reexcitation during phase 3 of the action potential. In addition to the factors discussed previously, the time-dependent changes in current are also crucial.
Refractoriness
Refractoriness describes the ability of a cell to respond to repetitive stimulation. This property is important for the normal function of the heart and is a determinant of susceptibility to arrhythmias. Stimulation during phase 2 results in no response, and the cell is said to be absolutely refractory. Stimulation during the middle and late parts of phase 3 may result in a response. However, during most of phase 3, the stimulus intensity has to be increased. There is a brief period in phase 3, the supernormal period, when the required stimulus is actually less than that during phase 4. For reexcitation, repolarization has to occur to a sufficiently negative potential that the Na+ current required to support a response can be activated. As the recovery of the Na+ channel from inactivation is not instantaneous, the available Na+ current at each potential during phase 3 is less than that predicted by the steady-state inactivation curve. The time interval between phase 0 of the action potential and the time that a second propagated response can be initiated is the effective refractory period of the cell. The effective refractory period of the cell is one of the determinants of the minimum path length for reentry.
The recovery of normal excitability closely parallels the APD. The refractory period may be prolonged by increasing the APD or by slowing the rate of recovery of the Na+ channel from inactivation. Class I antiarrhythmic drugs dissociate from the Na+ channel slowly, and, as a result, they decrease the fraction of available Na+ channels during phases 3 and 4. They also prolong refractoriness on the basis of the change in Na+ channel kinetics.
Conduction
For effective contraction, atria and ventricles have to be activated synchronously. The successful propagation of the cardiac impulse depends on the coupling of current sources and sinks. An action potential at a given site acts as a current source to excite neighboring cells (the current sinks). Large inward currents during phase 0 favor rapid propagation. Reduction of the magnitude of the inward current (e.g., by membrane depolarization and Na+ channel blocking drugs) slows conduction. Propagation from a current source occurs by the spread of local circuit currents between neighboring cells. The pathway for current spread is illustrated in Figure 57.4 and includes the coupling impedance between cells and the impedance of the sarcolemma; the extracellular fluid local circuit current discharges the capacity of the membrane of neighboring cells, thus bringing their potential to threshold. Low intercellular impedance favors rapid conduction. Specialized intercellular connections termed nexuses or gap junctions are the preferential sites for intercellular current. The gap junctions are formed by a family of proteins, the connexins. They aggregate at the sites of intercellular contacts such as the intercalated disks. Units from each of two neighboring cells associate to form fluid-filled channels. These channels have a gating mechanism that is controlled by intracellular pH and [Ca2+]i. Connexin expression is low in the SA and AV nodes and may be reduced during myocardial ischemia.
Until relatively recently, propagation in cardiac muscle was analyzed using a theory developed for current spread in homogeneous medium. Spach and associates (16,17) emphasized that at both the microscopic and macroscopic levels, cardiac muscle is not a homogeneous excitable medium. The low-resistance gap junctions are not uniformly distributed over the cell surface membrane. Gap junction density is greatest in the direction of fiber orientation. Hence, intercellular impedance is less in the longitudinal than in the transverse direction. With increasing age, connective tissue septa are laid down between fiber bundles and further distort the cardiac architecture. These structural changes may play a crucial role in the genesis of arrhythmias and are discussed in more detail in the section on reentry later in this chapter.
Changes in Ionic Currents during Pathologic States
Certain clinical states such as myocardial ischemia or reperfusion, cardiac failure, and myocardial hypertrophy are associated with an increased incidence of cardiac arrhythmias. Because the currents that generate normal action potential play a central role in most arrhythmias, these clinical states must exert some effect on the membrane currents. Membrane ion currents
can be modified in relatively few ways. Pathologic states may modify channel function by changing the transmembrane gradients for the permeant ions; they may alter channel gating by changing intracellular Ca2+, intracellular and extracellular pH, intracellular [ATP], and autonomic tone; and they may alter the level of expression of ion channels.
can be modified in relatively few ways. Pathologic states may modify channel function by changing the transmembrane gradients for the permeant ions; they may alter channel gating by changing intracellular Ca2+, intracellular and extracellular pH, intracellular [ATP], and autonomic tone; and they may alter the level of expression of ion channels.
Myocardial ischemia is associated with an early rise in [K+]o, a fall in intracellular and extracellular pH and [ATP], and an increase in [Ca2+]i and [Na+]i. Lysophospholipids also accumulate in the ischemic tissues. Elevation of [K+]o has two major effects: membrane depolarization and an increase in the inward rectifier K+ current. The fall in intracellular [ATP] and the associated rise in ADP activates the ATP-sensitive K+ channel. In general, most ionic currents are reduced by a fall in intracellular and extracellular pH. The threshold for this effect is variable between ion channels. The lysophospholipids, such as lysophosphatidylcholine, block ion channels. They may also change channel gating (e.g., inactivation of the Na+ current is slowed) (18). In experimental models of acute ischemia, reperfusion is associated with a high incidence of ventricular tachycardia and ventricular fibrillation. Indeed, the prevalence of arrhythmias during reperfusion may exceed that during the proceeding ischemic period. In the clinical setting, vasospastic angina and percutaneous transluminal coronary angioplasty are associated with reperfusion after a variable period of ischemia. Accelerated idioventricular rhythm is the most common arrhythmia in clinical reperfusion; ventricular fibrillation has a much lower prevalence than in the experimental setting. Reperfusion is associated with a restoration of [K+]o, pH toward normal, and generation of oxygen-derived free radicals. [Ca2+]i may rise further and reduce the conductance of junctional channels.
Cardiac hypertrophy and failure are associated with a prolongation of the action potential. The most consistent finding is a reduction of the transient outward current. Available data suggest that the decrease in Ito is the result of decreased expression of the channel. The mediators of this suppression are not known. Cytokines may play a role.
Electrophysiologic Mechanisms of Cardiac Arrhythmias
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


