50 More than a decade ago, the discovery and characterization of a mutation in the gene encoding a human cardiac voltage-gated sodium channel (SCN5A) began a new era in our understanding of the molecular and genetic basis of arrhythmias. Subsequent work has revealed an unexpectedly large number of clinically and pathophysiologically diverse conditions associated with an ever growing number of mutations in this gene (Box 50-1). The diversity of clinical disorders (phenotypes) associated with SCN5A mutations is explained in part by corresponding heterogeneity in mutant channel dysfunction. An understanding of the functional consequences of SCN5A mutations has driven tremendous progress in elucidating arrhythmia mechanisms in these genetic disorders. SCN5A encodes the pore-forming subunit of the major voltage-gated sodium channel expressed in human heart. This gene spans approximately 100 kilobases on the short arm of chromosome 3 (3p21) and comprises 28 canonical exons ranging in size from 53 (exon 24) to 3257 (exon 28) base pairs (Figure 50-1, A). The full-length product of SCN5A is a 2016 amino acid protein designated as NaV1.5, but other nomenclature (i.e., hH1) is also used to describe recombinant forms. Alternatively spliced messenger RNAs (mRNAs) transcribed from SCN5A have also been detected in heart.1 One splice variant expressed in adult heart is generated by alternative usage of splice acceptor sequences at the junction between intron 17 and exon 18, resulting in inclusion or exclusion of glutamine at position 1077.2 Approximately half of mature SCN5A mRNAs in heart encode the 2015 amino acid alternative form of the protein that arises from this alternative splicing event, and this impacts the functional properties of certain genetic variants.3,4 In fetal and neonatal heart, another alternatively spliced form of NaV1.5 is expressed that uses an alternative exon 6 (exon 6a; see Figure 50-1, A), resulting in several amino acid differences within a voltage-sensor domain (D1/S3-S4).1,5 This fetal-expressed NaV1.5 splice variant exhibits developmental regulation in mouse and human heart6 and shows distinct biophysical properties, including a more depolarized voltage dependence of activation. Other alternative splicing events have less certain biological and physiological relevance.1 Figure 50-1 SCN5A gene structure and mutations Transcription of human SCN5A is under the control of a promoter that precedes the first exon and other determinants of transcriptional activity located in the first intron.7 Common polymorphisms in the SCN5A promoter influence the level of gene expression and may have clinical relevance. In particular, a combination of polymorphisms constituting a common haplotype within the promoter has been associated with longer PR interval and QRS duration, as well as the extent of QRS widening during challenge with sodium channel blocking drugs in Asians.8 These and other observations suggest that unequal transcription from the two copies of SCN5A could influence the clinical expression of heterozygous mutations in this gene.9 The cardiac sodium channel resides in the myocyte plasma membrane as a multi-protein complex consisting of the pore-forming (α) subunit (NaV1.5), auxiliary (β) subunits, and other interacting proteins. A family of four sodium channel β-subunits (β1, β2, β3, β4) encoded by the genes SCN1B, SCN2B, SCN3B, and SCN4B are expressed in heart and likely interact with NaV1.5, possibly to modulate channel function or subcellular localization.10 Several other proteins expressed in heart also interact directly or indirectly with sodium channels, including ankyrins, caveolin-3, calmodulin, α1-syntrophin, glycerol-3-phosphate dehydrogenase 1-like protein (GPD1L), fibroblast growth factor homologous factors, Nedd4-like ubiquitin-protein ligase, multi-copy suppressor of gsp1 (MOG1), and 14-3-3η.11 Some of these proteins have been implicated in rare cases of inherited arrhythmia susceptibility, suggesting that they mediate important functional interactions.12 Further information about regulation of sodium channels in the context of multi-protein complexes is provided in Chapter 18. Cardiac voltage-gated sodium channels are critical mediators of phase 0 depolarization and control the velocity of impulse propagation through the heart. Sodium channels switch between three major functional states (closed, open, and inactivated) in response to changes in membrane potential. Only open channels generate electrical current by allowing the selective passage of sodium ions into cells. The functional state of sodium channels can be monitored in electrophysiological recording experiments by using specific voltage-clamp protocols to elicit activation (closed → open), inactivation (open → inactivated), and recovery from inactivation (inactivated → closed), which occur on a millisecond time scale. In addition to these rapid transitions, sodium channels are susceptible to slow inactivation if the membrane remains depolarized for a longer time. Slow inactivation occurs on a time course of several seconds and may contribute to determining the availability of active channels under various physiological conditions. The structural basis for sodium channel function is discussed in Chapter 1, and additional details about functional properties of sodium channels are presented in Chapter 9. More than 450 SCN5A mutations have been identified in patients with the arrhythmia predisposition syndromes presented in Box 50-1. Types of mutations include missense (e.g., amino acid substitutions), nonsense (e.g., premature stop codon), insertions or deletions, and splice-site mutations. Some mutation types, particularly those predicted to truncate the encoded protein (nonsense, insertions, or deletions affecting the reading frame), destroy the functionality of the channel (loss-of-function) by deleting critical domains or preventing protein translation through nonsense-mediated decay of the mRNA. Splice-site mutations may also severely disrupt channel function if exons are skipped or intron sequences are retained in the final mRNA. However, a large number of SCN5A mutations result in single amino acid substitutions for which functional predictions are difficult. Several missense mutations and a few in-frame insertion/deletion alleles have been studied in vitro to determine the molecular basis for arrhythmia predisposition in these syndromes, and common patterns of sodium channel dysfunction have emerged. Congenital long QT syndrome (LQTS) is an inherited condition of abnormal myocardial repolarization. It is characterized clinically by increased risk of potentially fatal ventricular arrhythmias, especially torsades de pointes, manifesting as syncope, cardiac arrest, and sudden unexplained death in otherwise healthy young adults and children. The disease is typically recognized in late childhood or early adolescence, but extreme cases may present during early infancy or in the perinatal period.13 The syndrome most often transmitted is in families as an autosomal dominant trait (Romano-Ward syndrome; see also Chapter 93). Approximately 10% of autosomal dominant LQTS cases are explained by SCN5A mutations,14 and this form of the disease is referred to as LQT-3. The proportion of sodium channel mutations in LQTS cases presenting during the perinatal and neonatal time periods may be higher.15 The first SCN5A mutation described was an in-frame deletion of three highly conserved amino acid residues (delKPQ 1505-1507) located within the cytoplasmic loop connecting domains 3 and 4 of NaV1.516—a structural domain required for fast inactivation of the channel. Since the discovery of the first SCN5A mutation, more than 75 mostly missense SCN5A mutations have been associated with LQT-3 (Figure 50-1, B). Although mutations are found throughout the channel sequence, multiple mutations are clustered in a small number of structural regions. One of these regions is the S4 segment of domain 4 (D4), a critical structural determinant of voltage sensing. Another mutation cluster can be found in the aforementioned cytoplasmic inactivation gate. Finally, several mutations are found in the proximal carboxy terminus, which includes binding sites for several interacting proteins that can modulate sodium channel function.11 Persons with SCN5A mutations associated with LQTS often present with distinct clinical features, including sinus bradycardia, and a tendency for cardiac events to occur during sleep or rest.17 In addition to bradycardia, certain features of the surface electrocardiogram (ECG), such as narrow and late-onset peaked or biphasic T waves, may offer additional clues to the presence of an SCN5A mutation in the setting of LQTS.18 Cardiac events are less frequent in the settings of SCN5A mutations as compared with the two major forms of LQTS caused by potassium channel mutations (LQT-1, LQT-2).19,20 However, in children and adults, the risk of dying after a cardiac event is greater for LQT-3 than for either LQT-1 or LQT-2. In LQT-3, most SCN5A mutations exhibit a dominant gain-of-function phenotype at the molecular level characterized by defective inactivation leading to increased persistent sodium current (Figure 50-2).21 Some mutant channels may also exhibit accelerated recovery from inactivation—a phenomenon consistent with an unstable inactivated state. At the level of single sodium channels, increased persistent sodium current has been correlated with an increased tendency for channel reopening, which may occur in bursts.21 More severe slowing of inactivation may be noted in mutations associated with clinically severe LQTS,13,22 while a small number of mutations may alter voltage dependence of activation and/or inactivation with less dramatic effects on the level of persistent current.23,24 Figure 50-2 Increased persistent sodium current in LQT-3 Increased persistent sodium current has also been observed when wild type NaV1.5 channels were coexpressed with LQTS-associated mutations in certain auxiliary subunits and interacting proteins. Specifically, mutations in SCN4B encoding the β4 auxiliary subunit,25 CAV3 encoding the vesicular trafficking protein caveolin-3,26 and SNTA1 encoding the adapter protein α1-syntrophin,27,28 exert their pathologic effects by disturbing sodium channel inactivation, leading to increased persistent current. The mechanism responsible for increased persistent current associated with SNTA1 mutations involves activation of neuronal nitric oxide synthase (nNOS) complexed with NaV1.5, leading to increased cell levels of nitric oxide and nitrosylation of the sodium channel. Activation of nNOS is the result of disrupted scaffolding of a plasma membrane Ca2+-ATPase (PMCA4b) that normally inhibits the enzyme in a multi-protein complex with NaV1.5 and α1-syntrophin.28 Increased persistent sodium current provides an explanation for delayed repolarization in LQTS.29 Cardiac action potentials last several hundred milliseconds because of a prolonged depolarization phase (plateau), the result of opposing inward (mainly Na+ and Ca2+) and outward (K+) ionic currents. Repolarization occurs when net outward current exceeds net inward current. Increased persistent sodium current will shift this balance toward inward current and will delay onset of repolarization, thus lengthening the action potential duration and the corresponding QT interval. Delayed repolarization predisposes to ventricular arrhythmias mainly by increasing the probability of early afterdepolarization (EAD) and by increasing the dispersion of the action potential duration.30 These phenomena promote conditions that allow electrical signals from depolarized regions of the heart to prematurely reexcite adjacent myocardium that has already repolarized—the basis for a reentrant arrhythmia. The pathogenic effects of increased persistent sodium current in LQTS have also been strongly supported by computer simulations of cardiac action potentials29,31 and by electrophysiological investigations of cardiac myocytes from genetically engineered mice. Mice engineered to carry the delKPQ mutation have spontaneous life-threatening ventricular arrhythmias.32,33 At the cellular level, cardiomyocytes from these LQT-3 mice exhibit prolonged action potential duration and frequent EADs—findings that were exaggerated by slow pacing rates. These cells have increased persistent sodium current with faster recovery from inactivation—features that were predicted from studies performed in noncardiac cells.32 Further evidence from the delKPQ mouse suggests that delayed afterdepolarization (DAD) caused by a Ca2+-dependent diastolic transient inward current, possibly evoked by abnormal Na+ entry through mutant channels, also contributes to arrhythmia susceptibility and may account for the greater lethality of this LQTS subtype.34 Knowledge of the basic mechanisms underlying LQT-3 has prompted new ideas regarding genotype-specific treatment. Mexiletine and other sodium channel blocking drugs can reduce persistent sodium current and shorten the QT interval in LQT-3 patients,35–38 although no data yet indicate that this therapeutic strategy will reduce mortality. In LQT-3 mouse models, mexiletine shortens the myocyte action potential and prevents arrhythmias,30,39 mainly during slow pacing rates. Suppression of increased persistent current by the antianginal drug ranolazine has been demonstrated for LQT-3 mutations in vitro,40 and ranolazine treatment of human with carrying the SCN5A-delKPQ mutation shortened the QTc interval significantly.41 Propranolol, but not the other β-adrenergic antagonists, may exhibit sodium channel blocking effects at high concentrations,42–44 and certain SCN5A mutations have increased sensitivity to the drug.13 However, propranolol and other β-blockers did not suppress arrhythmias in the delKPQ mouse model.33 SCN5A mutations may cause life-threatening LQTS during gestation (fetal LQTS) and in neonates (neonatal LQTS). Clinical signs suggestive of fetal LQTS include ventricular tachycardia, second-degree atrioventricular (AV) block, and, most commonly, sinus bradycardia,45 but such findings may go undetected because electrocardiographic testing of fetuses is not routine. Certain SCN5A mutations, many of which are de novo,13,46–49 present with earlier onset and more severe congenital arrhythmia syndromes than is typical for LQT-3. The high rate of reported de novo SCN5A mutations in the perinatal period may reflect low heritability due to a survival disadvantage conferred by severe, life-threatening phenotypes. The functional effects of certain SCN5A mutations associated with fetal LQTS can also be potentiated by a developmentally regulated splice isoform involving alternative forms of exon 6.6 Evidence indicates that occult LQTS can clinically present as sudden infant death syndrome (SIDS) (see Chapter 98). Cardiac mechanisms including life-threatening arrhythmias have been suspected as risk factors for SIDS, and mutations in genes responsible for LQTS have been identified in approximately 10% of cases.50,51 SCN5A mutations have accounted for approximately 50% of all LQTS mutations identified in SIDS. SCN5A mutations associated with SIDS promote increased persistent current overtly or under certain conditions such as intracellular acidosis or in the context of a common splice variant.50,52–54 Common variants within the SCN5A coding region have been identified in certain populations, and some of these may confer increased risk of cardiac arrhythmia. One variant common in subjects of African descent (S1103Y, also reported as S1102Y) has been associated with an eightfold increased risk for ventricular arrhythmia.55 Functional consequences of S1103Y include increased transient and increased persistent current sufficient to evoke EADs in a computational model of cardiac action potentials. This variant may also increase arrhythmia risk in infants. One study suggested that African American SIDS victims are more often homozygous for SCN5A-S1103Y when compared with non-SIDS infants.53 This study further demonstrated that SCN5A-S1103Y channels exhibit a greater level of persistent sodium current when exposed to intracellular acidosis. A subsequent study reported a significantly higher prevalence of heterozygous SCN5A-S1103Y carriers among 71 African American cases of SIDS as compared with African American controls.56 Other common SCN5A variants may also be proarrhythmic because of altered sodium channel inactivation. The common variant R1193Q is common in Asians but is a rare variant (0.3%) in subjects of European ancestry.57 This variant has been reported in subjects with both acquired and congenital LQTS58,59 and in sudden unexplained death syndrome (SUDS).60 This variant promotes an increased persistent sodium current58 and faster inactivation.60 Mutations in SCN5A have been associated with 20% to 30% of cases of Brugada syndrome (BrS), a heritable form of idiopathic ventricular fibrillation.61,62 Individuals with BrS have increased risk for potentially lethal ventricular arrhythmias (polymorphic ventricular tachycardia or fibrillation) typically during sleep, but in the absence of myocardial ischemia, electrolyte abnormalities, or structural heart disease. Increased risk for atrial fibrillation and intraventricular conduction abnormalities may also occur in BrS. Individuals with the disease may exhibit a characteristic baseline ECG pattern consisting of ST elevation in the right precordial leads but normal QT intervals.63 Administration of sodium channel blocking agents (e.g., procainamide, flecainide, ajmaline) or fever may expose this ECG pattern in latent cases.64,65 Inheritance is autosomal dominant with incomplete, often low, penetrance and a substantial male predominance. A family history of unexplained sudden death is typical. The sudden unexplained death syndrome (SUDS) is clinically similar to BrS and causes sudden death, typically during sleep, in young and middle-aged males in Southeast Asian countries.66 Additional information about BrS may be found in Chapter 92. Chen et al first identified three distinct SCN5A mutations in two unrelated BrS families and in a third sporadic case.61 One mutation was missense (T1620M), and the other alleles included a frameshift caused by a single nucleotide deletion and a putative splice-site defect. More than 375 mutations have been reported in BrS (Figure 50-1, C, D). SCN5A mutations have also been identified in subjects with SUDS.60
Mechanisms in Heritable Sodium Channel Diseases
Cardiac Sodium Channel
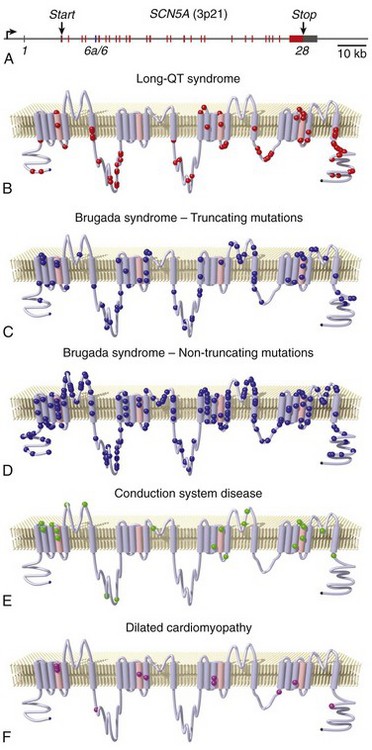
A, Schematic of the SCN5A gene illustrating 28 canonical exons and one alternative exon (vertical lines; red for canonical coding exons, blue for alternative exon 6a, and dark gray for noncoding exons or portions of exons). The bent arrow indicates the approximate location of the transcription start site near the promoter. Locations of the translation start and stop codons are indicated. Locations of mutations associated with LQT-3 (B), BrS (C,D), cardiac conduction disease (E), and dilated cardiomyopathy (F) are superimposed on a two-dimensional membrane topology map of the sodium channel protein (Mutation data were obtained from the online Inherited Arrhythmias Database [http://www.fsm.it/cardmoc/] and from the literature. Images were prepared by Dr. Robert Abraham, Vanderbilt University, using software generously supplied by Dr. André Linnenbank, University of Amsterdam.)
Normal Sodium Channel Function
Consequences of Sodium Channel Dysfunction
Delayed Repolarization
Congenital Long QT Syndrome
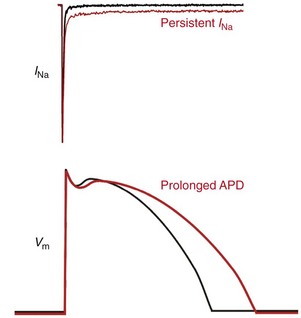
Upper panel is a representative current recording from a cell expressing either wild type (WT) cardiac sodium channels (black trace) or an SCN5A mutation associated with LQT-3 (red trace) illustrating increased persistent sodium current (INa). Lower panel illustrates a normal ventricular action potential waveform (black line) and an illustration of prolonged action potential duration (APD; red line) as occurs in LQTS.
Perinatal and Neonatal LQT-3
Arrhythmia Susceptibility With Common SCN5A Variants
Impaired Impulse Propagation
Brugada Syndrome
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Mechanisms in Heritable Sodium Channel Diseases
