The primary cause of morbidity and mortality in patients with Marfan syndrome (MFS) is related to the propensity to aortic aneurysm formation and associated dissection. Noninvasive cardiovascular imaging has contributed to the improved survival noted among patients with MFS in the current era. Ascending aortic dilatation is usually readily detected with transthoracic echocardiography (TTE) and transesophageal echocardiography (TEE). Echocardiography is a very important imaging tool in the diagnosis and management of patients with MFS. In addition, both computed tomography angiography (CTA) and magnetic resonance imaging (MRI) play an important role in the diagnosis, management, and surgical planning for patients with MFS and other thoracic aortic diseases.
MFS, an autosomal dominant multisystem disorder, is the most common systemic connective tissue disease, occurring in 2 to 3 per 10,000 individuals. Cardiovascular complications occur in the majority of patients. Classically MFS is caused by a mutation of the fibrillin-1 gene (FBN1), which maps to 15q21. FBN1 mutations increase the susceptibility of fibrillin, a structural protein, to proteolysis in vitro leading to fragmentation of microfibrils. Fragmentation of the elastic fibers in the aortic media is a histological marker of MFS, so-called medial degeneration. Transforming growth factor beta (TGFβ), a regulatory protein, has been shown to bind to fibrillin in microfibrils. The proteolysis of fibrillin in MFS results in increased bioavailability of TGFβ, which in turn causes excess signaling and ultimately leads to abnormal elastic properties that make the aorta stiffer and less distensible than normal.
More than 500 different mutations involving the FBN1 gene have been identified to date, and the penetrance of the fibrillin mutation is high. However, no correlation has been recognized between the specific type of FBN1 mutation and the clinical phenotype. In approximately 75% of cases, an individual with MFS inherits the disorder from an affected parent, the remaining 25% result from de novo mutation. Genetic counseling should be provided at the time of initial evaluation or diagnosis, as well as to potential parents.
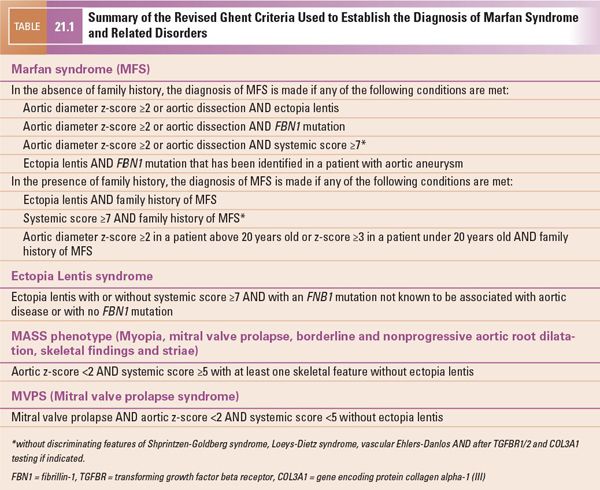
The revised Ghent diagnostic criteria were published in 2010 (Table 21.1). These criteria give more weight to the cardinal features of MFS, including aortic root aneurysm or dissection and ectopia lentis. They also establish a more prominent role for the use of genetic testing. The involvement of other organ systems contributes to a “systemic score” that is used when the patient does not meet criteria based on aortic disease, ectopia lentis, family history, or FBN1 mutation (Table 21.2). Confirmation of the diagnosis of MFS requires a complete personal and family history and a comprehensive multidisciplinary approach involving genetic, cardiac, ophthalmologic, and, in select cases, orthopedic consultations and various diagnostic tests.
ANATOMY AND PHYSIOLOGY OF THE CARDIAC LESION
The cardiovascular features of MFS were initially reported by McKusick et al. Dilatation of the ascending aorta at the level of the aortic sinuses, also known as the aortic root, is the most common and characteristic cardiovascular manifestation of MFS (Fig. 21.1). Progressive aortic sinus enlargement is present in approximately 50% of adults and children with MFS.
Ascending aortic aneurysm in MFS can be readily detected by TTE in most patients, and serial TTE studies can usually be performed to monitor the size of the dilated ascending aorta. Alternate imaging modalities such as TEE (Fig. 21.2), CTA (Fig. 21.3), or MRI (Fig. 21.4) are used to confirm the diameter of the ascending aorta and to completely assess the arch, descending thoracic, and abdominal aorta, which are often incompletely visualized on TTE. Aortic dissection (Fig. 21.5) or rupture accounts for most of the premature mortality in patients with MFS. Marked improvement in life expectancy in patients with MFS has been noted over the past 30 years. This change in prognosis is largely the result of early diagnosis of MFS, initiation of medical therapy, and serial aortic imaging with recognition of aortic aneurysmal disease and prophylactic aortic root replacement. Thus, it is critically important to consider the diagnosis and to perform serial cardiovascular imaging studies in all patients with confirmed or suspected MFS.
Aortic dissection data suggest that MFS is present in 50% of patients presenting with aortic dissection who are younger than 40 years of age. Risk factors for aortic dissection in MFS include (a) aortic sinus diameter greater than 50 mm, (b) aortic dilatation extending beyond the sinuses of Valsalva, (c) more than 5% per year increase in aortic size in children, or an increase of more than 5 mm per year in adults, and (d) family history of aortic dissection. Aortic diameter should be measured at multiple levels by TTE and compared with normal values based on age and body surface area. The aortic root diameter increases with age and is larger in men than women (Fig. 21.6). Serial measurements are indicated. When adequate assessment of the aorta is not possible by TTE, alternate imaging modalities are used such as CTA, MRI, or TEE.
Patients with MFS are predisposed to dilatation or dissection of the descending thoracic aorta, although this is less common than involvement of the ascending aorta. Complete evaluation of the distal ascending aorta, arch, and descending thoracic aorta is often challenging in adults, and CTA or MRI play an important role in evaluation of the entire aorta (Fig. 21.7). Dilatation or dissection of the descending thoracic aorta is a recognized cardiovascular complication of MFS, and can be evaluated with CTA (Fig. 21.8), TEE (Fig. 21.9, Videos 21.1 and 21.2), or MRI. Nevertheless, life-long imaging of the entire aorta is essential for the optimal management of patients with MFS.
Important cardiovascular manifestations in MFS other than aortic dilatation have been recognized. Mitral valve prolapse (Fig. 21.10) occurs in approximately 60% of patients with MFS. The mitral valve leaflets are longer and thinner, with less posterior leaflet prolapse and more anterior or bileaflet prolapse compared with mitral valve prolapse in non-MFS patients. In addition, patients with MFS develop severe degrees of mitral valve regurgitation and present for surgery at a younger age than non-MFS patients. Less common cardiovascular complications of MFS include mitral annular calcification, tricuspid valve prolapse, and pulmonary artery dilatation in the absence of pulmonary valve disease. These less common complications have been removed from the diagnostic criteria because they are less specific for MFS. Central aortic valve regurgitation caused by annular enlargement occurs as the aorta enlarges (Fig. 21.11, Videos 21.3 and 21.4). There are reports of left ventricular dilatation and systolic dysfunction, regardless of valvular regurgitation in patients with MFS, and other reports suggesting that ventricular dysfunction is uncommon in the absence of valve disease.
CLINICAL PRESENTATION
Despite the progress in management of patients with MFS and sophistication of the health care system, patients with MFS still die of aortic dissection before consideration and diagnosis of the disorder. In retrospect, many of these patients have physical features or a family history of MFS that should have prompted cardiovascular screening before the aortic catastrophe.
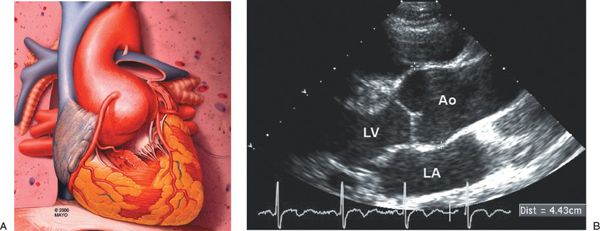
Figure 21.1. Aortic Enlargement in Marfan Syndrome. A: Schematic of the typical aortic enlargement in Marfan syndrome. This schematic demonstrates enlargement of the proximal aorta at the level of the aortic sinuses. B: Two-dimensional echocardiogram in the parasternal long-axis image orientation demonstrates dilatation of the ascending aorta at the level of the aortic sinuses. The electronic calipers demonstrate the leading edge–to–leading edge method for ascending aortic measurement. Ao, aorta; LA, left atrium; LV, left ventricle.
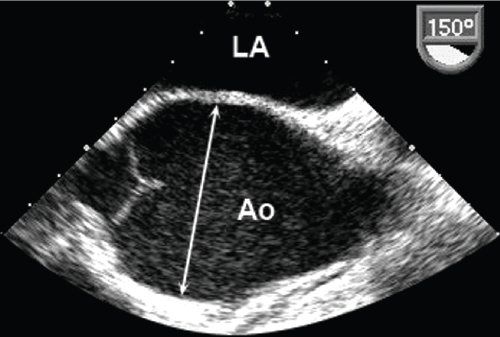
Figure 21.2. Transesophageal echocardiogram in the longitudinal image plane at 150 degrees. Dilatation of the ascending aorta at the level of the aortic sinuses (arrow). Ao, aorta; LA, left atrium.
The majority of patients with MFS have a family history of the disorder and are identified during routine family echocardiographic screening. Skeletal, ocular, and occasionally pulmonary features may prompt cardiovascular screening of the patient and family. When a patient is initially diagnosed with MFS, screening of all first-degree relatives is recommended. When MFS is suspected, screening with TTE is recommended to identify cardiovascular disease—most importantly, aortic aneurysmal disease.
Marfan Syndrome in Children
The revised Ghent diagnostic criteria address the issue of making the diagnosis of MFS in children. Previously, this was challenging as the features may be subtle early in life and develop with age. Based on the revised criteria, if an individual <20 years old with suspected MFS does not meet diagnostic criteria as described in Table 21.1, specific recommendations are given based on the findings present. When the systemic score is <7 and/or there is borderline aortic root enlargement (z-score <3) in a patient with no FBN1 mutation and no family history, the term “nonspecific connective tissue disorder” is recommended until follow-up evaluation demonstrates aortic root z-score ≥3. If an FBN1 mutation is identified in a young patient with z-score <3, the term “potential MFS” is used until the aortic root z-score is ≥3. Children with suspected MFS should have comprehensive evaluation during preschool, before puberty, and at age 18 years, because some of the clinical manifestations of MFS become evident with time. Annual aortic imaging follow-up is recommended for children with MFS or when the aorta is enlarged regardless of diagnostic criteria.
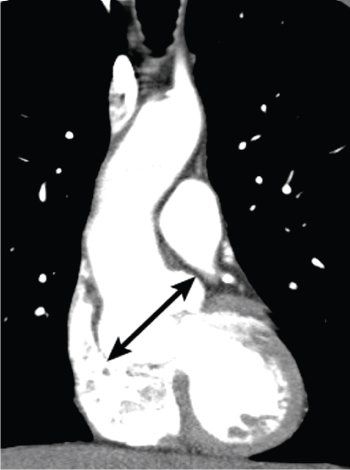
Figure 21.3. Computed tomography (CT) examination of the chest using a dual-source CT scanner with intravenous contrast material. Reformatted in the coronal plane, shows marked dilatation of the ascending aorta at the level of the aortic sinuses (arrow) in a patient with Marfan syndrome. Note that the rest of the aorta is near normal caliber.
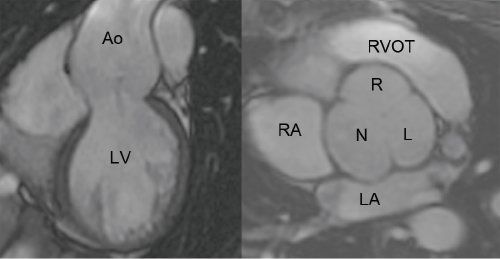
Figure 21.4. Electrocardiographic-gated, steady state free precession magnetic resonance imaging. The coronal plane (left) and the double oblique plane through the sinus of Valsalva (right) demonstrate dilatation of the aortic root and a tricuspid aortic valve. Ao, aorta; LV, left ventricle; RA, right atrium; LA, left atrium; N, noncoronary cusp; R, right coronary cusp; L, left coronary cusp; RVOT, right ventricular outflow tract.
The majority of patients with MFS demonstrate aortic root dilatation, mitral valve prolapse, or both before age 18 years. It is important to make the diagnosis and to initiate appropriate medical therapy and serial screening in an effort to prevent or slow aortic enlargement and thus delay aortic operative intervention.
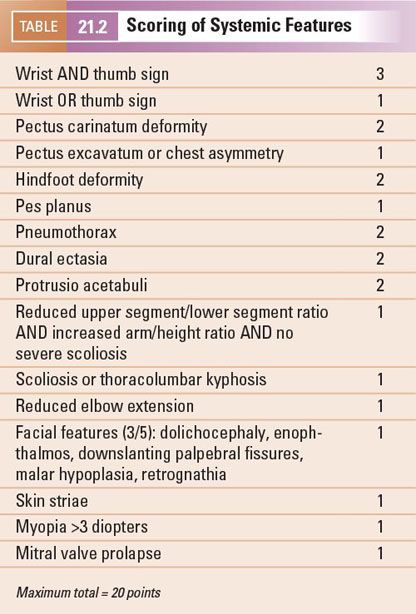
Beta-blockers have been demonstrated to significantly decrease the rate of aortic dilatation at the level of the aortic sinuses in children and adults and therefore should be prescribed either at the time of diagnosis or on documentation of aortic enlargement. Angiotensin receptor blockers (ARBs) have been shown to be beneficial in slowing the rate of aortic dilatation in a small cohort of MFS children who demonstrated progressive aortic dilatation on beta-blocker therapy. Multiple trials are ongoing comparing the use of beta-blockers and ARBs in MFS patients with aortic enlargement.
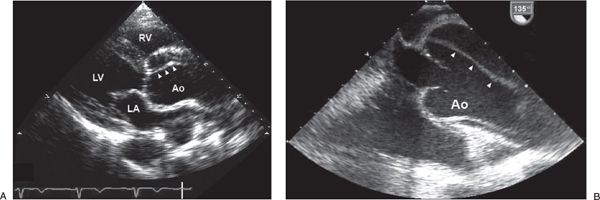
Figure 21.5. Aortic Dissection. A: Two-dimensional transthoracic echocardiogram in the parasternal long-axis image orientation showing dilatation of the ascending aorta and proximal aortic dissection (arrowheads). B: Transesophageal echocardiogram in a longitudinal image orientation showing dilatation of the ascending aorta and proximal aortic dissection (arrowheads). The transesophageal echocardiogram was performed in the operating room before surgical repair. Ao, aorta; LA, left atrium; LV, left ventricle; RV, right ventricle.
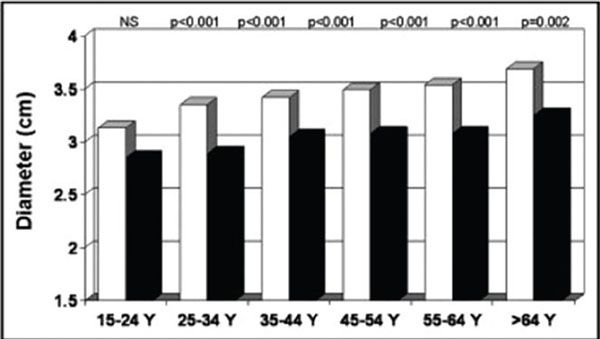
Figure 21.6. Mean aortic root diameter adjusted for body surface area in men (white bars) and women (black bars) by decade of age. (From Devereux et al., Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons ≥15 years of age, Am J Cardiol. 2012;110[8]:1189–1194.)
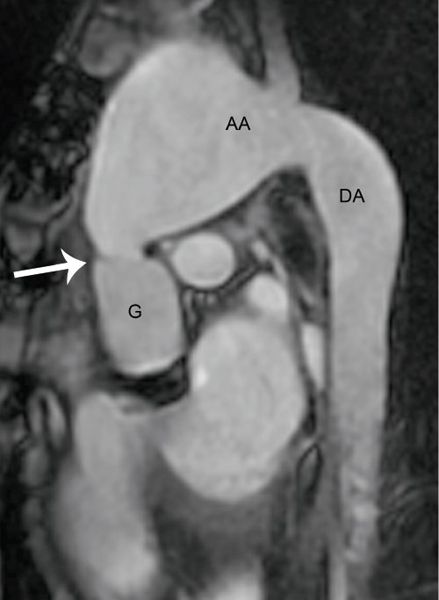
Figure 21.7. MRI in a patient with Marfan syndrome and prior composite root replacement. Steady state free precession imaging in an oblique sagittal plane demonstrates severe dilatation of the distal native ascending aorta and arch beginning at the distal anastomosis from the prior aortic root replacement operation (arrow). The proximal descending thoracic aorta is also mildly dilated. G, graft; AA, aortic arch; DA, descending aorta.
Neonatal MFS is a severe form of MFS apparent at birth that carries a poor prognosis. Aortic enlargement with associated severe aortic valve regurgitation is often present. In addition, progressive mitral and/or tricuspid valve prolapse with regurgitation leading to congestive heart failure is common and affects management and patient survival. Characteristic noncardiac features include infantile pulmonary emphysema, ectopia lentis, arachnodactyly, joint contractures, and loose skin.
CARDIAC COMPLICATIONS IN MARFAN SYNDROME
The most important, and life-threatening, cardiovascular complication in MFS is aortic dissection (see Figs. 21.8 and 21.9, Videos 21.1 and 21.2) or rupture. This most commonly involves the ascending aorta but can also affect the descending thoracic aorta. Additional cardiovascular complications include progressive valvular regurgitation, such as aortic regurgitation caused by annular enlargement or mitral or tricuspid valve regurgitation caused by leaflet prolapse.
Prophylactic beta-blockade has been demonstrated to be effective in slowing the rate of aortic dilatation and reducing the development of aortic complications. These medications are generally recommended in patients with MFS. The ARB losartan has been demonstrated to have an important impact on vascular development in the mouse model of MFS. ARBs also slowed the rate of aortic dilatation in a small cohort of children with MFS and studies comparing beta-blockers and ARBs are ongoing in both children and adults. ARBs should be considered as an alternative medication option for patients with MFS intolerant of beta-blockers.
Genetic counseling is recommended for all patients with MFS. Due to the autosomal dominant nature of the disorder, each offspring of an affected Marfan parent has a 50% chance of inheriting the genetic mutation. The risk of transmission to the fetus should be discussed before starting a family.
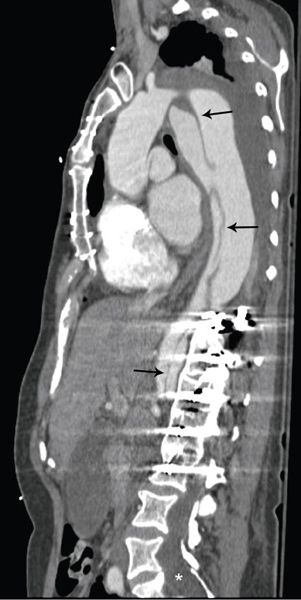
Figure 21.8. Descending thoracic aorta dissection in a patient with Marfan syndrome and prior composite root replacement. Computed tomography angiography in an oblique sagittal plane demonstrates a dissection originating in the arch and extending into the abdominal aorta (black arrows). Note the artifact in the thoracic spine from prior scoliosis repair and the sacral dural ectasia (white asterisk).
Cardiac Surgical Intervention
Aortic root replacement in patients with MFS is recommended when the ascending aorta reaches a diameter of 5 cm or more because of the increased risk of aortic rupture. Select patients are referred for aortic root replacement with an aortic dimension less than 5 cm; these include patients with a family history of aortic dissection, patients with a rapid rate of aortic dilatation (greater than 5% per year, or more than 5 mm per year in adults), those interested in future pregnancy with an aortic dimension greater than 40 to 45 mm, and those interested in the valve-sparing aortic root replacement (see Fig. 21.13). Alternatively, if the maximal cross-sectional area in square centimeters of the ascending aorta or root divided by the patient’s height in meters exceeds 10, prophylactic surgery can be recommended, as patients with a smaller body size may develop complications at an aortic diameter less than 5 cm. Patients with aneurysms measuring less than 5 cm and no high-risk characteristics require serial follow-up studies to measure the aortic dimensions and decide the appropriate timing of intervention. The surgical options include the Bentall composite aortic valve and ascending aorta replacement (Fig. 21.12) or the aortic valve-sparing root replacement (Fig. 21.13).
Cardiovascular surgery can also be safely performed in children with MFS. Indications for surgical intervention in the pediatric population include (a) rapid rate of growth of the ascending aorta (greater than 5 mm per year), (b) progressive aortic valve regurgitation, or (c) the need for mitral valve surgery in patients with marked aortic enlargement. The surgical options are the same as in adults and include composite graft repair (see Fig. 21.12), or the valve-sparing procedure (see Fig. 21.13). Both procedures have shown excellent results for prophylactic replacement of an enlarged aortic root in older children and adults.
Pregnancy in Patients with MFS
The risk of aortic dissection during pregnancy is increased in patients with MFS. This is the result of a combination of the preexisting medial aortic disease with superimposed hormonal inhibition of aortic collagen and elastin deposition, and the hyperdynamic, hypervolemic circulatory state of pregnancy. There is a reported 11% complication rate associated with pregnancy in patients with MFS, mostly related to aortic rupture and endocarditis. The overall risk of death during pregnancy in patients with MFS is around 1%.
Women with MFS are counseled against proceeding with pregnancy when the ascending aorta exceeds 40 to 45 mm, depending on concomitant risk factors including personal or family history of aortic dissection, and rapid aortic dilatation. The risk of aortic complication is increased during pregnancy in patients with MFS when the aortic root diameter exceeds 40 to 45 mm at the start of pregnancy, and the risk is further increased when the aorta dilates during pregnancy. The risk of dilatation of the aorta during pregnancy in the MFS patient has been reported to be lowest in the first trimester and greatest in the third trimester, as well as during labor and the early postpartum period. Beta-blocker therapy should be continued throughout pregnancy, and patients should have serial follow-up echocardiograms to assess the change in the size of the aorta during pregnancy. ARBs should not be used during pregnancy. The frequency of aortic imaging should be individualized. Aortic root replacement should be considered during pregnancy in patients with MFS with progressive aortic dilatation or for documented aortic dissection.
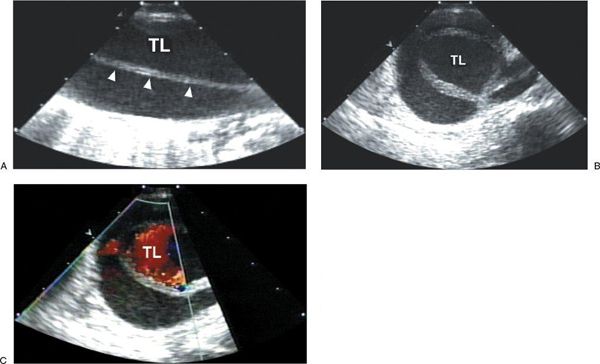
Figure 21.9. Descending thoracic aortic dissection demonstrated by transesophageal imaging. A: Longitudinal view of the descending thoracic aorta demonstrating dissection flap (arrowheads) with true lumen (TL) and false lumen. B: Transverse view of the descending thoracic aorta demonstrating dissection and true lumen (TL) and false lumen. C: Color flow imaging of the transverse view demonstrating primary flow in the TL of the dissected descending thoracic aorta.
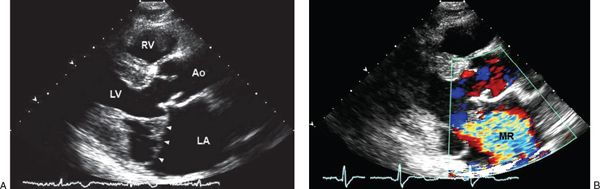
Figure 21.10. Mitral valve prolapse in Marfan syndrome. A: Bileaflet mitral valve prolapse (arrowheads) noted by two-dimensional echocardiographic parasternal long-axis imaging in a patient with Marfan syndrome. B: Associated with severe mitral regurgitation by color flow imaging. Note enlargement of the left atrium and aortic sinuses. Ao, aorta; LA, left atrium; LV, left ventricle; MR, mitral regurgitation; RV, right ventricle.
Assisted vaginal delivery can be considered in patients with MFS when the aortic root diameter is less than 40–45 mm, the aorta has not demonstrated change during pregnancy, and there is no associated severe cardiovascular disease. For patients with MFS with other characteristics, planned cesarean delivery is generally the preferred mode of delivery. Antibiotic prophylaxis administered around the time of delivery is appropriate for those patients with prior root and valve replacement surgery or a past history of endocarditis. Postpartum uterine hemorrhage is a common complication of MFS, occurring in nearly 40% of women. The risk of aortic dissection persists during the early postpartum period, thus these patients should be monitored postpartum.
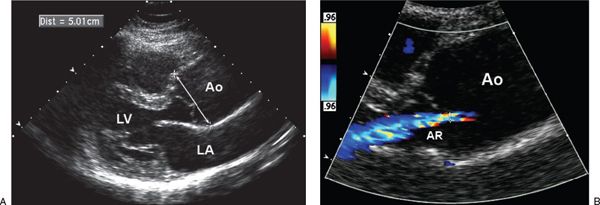
Figure 21.11. Aortic annular dilation and regurgitation. A: Parasternal long-axis image demonstrating aneurysmal enlargement of the ascending aorta (arrow). B: Color flow imaging demonstrating aortic valve regurgitation caused by annular dilatation. Ao, aorta; AR, aortic valve regurgitation; LA, left atrium; LV, left ventricle.
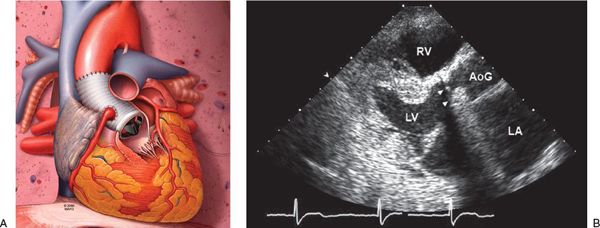
Figure 21.12. Bentall operation. A: Schematic of the Bentall operation. In this operative procedure, the proximal aorta is replaced with a valved conduit, and the coronary arteries are reimplanted. This schematic demonstrates a mechanical valve prosthesis. Biological prostheses can also be used. B: Transthoracic echocardiographic image of a patient with Marfan syndrome after a Bentall operation. The aortic valve and proximal ascending aorta are replaced with a mechanical valve prosthesis (arrowheads) and aortic graft, respectively. AoG, aortic graft; LA, left atrium; LV, left ventricle; RV, right ventricle.
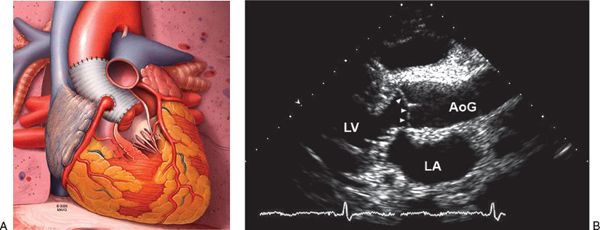
Figure 21.13. Valve-sparing aortic root replacement. A: Schematic of the valve-sparing aortic root replacement. The ascending aorta is replaced with a graft material, the native aortic valve is preserved, and the coronary arteries are reimplanted. B: Transthoracic echocardiographic image after a valve-sparing aortic root replacement operation. The native aortic valve remains (arrowheads, spared native aortic valve), and proximal ascending aorta is replaced with an aortic graft. AoG, aortic graft; LA, left atrium; LV, left ventricle.
BASICS OF ECHOCARDIOGRAPHIC ANATOMY AND IMAGING
Two-Dimensional Echocardiographic Anatomy and Hemodynamics
A comprehensive TTE examination will often demonstrate the cardiovascular features of MFS in the involved patient but may be normal or near normal despite a confirmed diagnosis of MFS.
Echocardiographic imaging of the aorta includes the parasternal long-axis view to measure the dimensions of the aortic sinuses, sinotubular junction, and ascending aorta. The leading edge–to–leading edge technique was previously used to measure the ascending aorta; however, the current recommendation is to measure the ascending aorta using an inner edge–to–inner edge technique. An off-axis parasternal image also demonstrates the descending thoracic aorta in many patients (Fig. 21.14). The aortic arch and descending thoracic aorta are imaged using the suprasternal window (Fig. 21.15), and the abdominal aorta is imaged from the subcostal format (Fig. 21.16). Aortic dimensions should be compared to normal for patient age and body surface area (see Fig. 21.6).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


