The safety and efficacy of an amlodipine/olmesartan medoxomil (OM)-based titration regimen was assessed in patients with type 2 diabetes mellitus and hypertension. After a 2- to 3-week placebo run-in period, 207 patients received amlodipine 5 mg and were uptitrated to amlodipine/OM 5/20, 5/40, and 10/40 mg and then amlodipine/OM 10/40 mg plus hydrochlorothiazide 12.5 and 25 mg in a step-wise manner at 3-week intervals if the seated blood pressure (BP) remained ≥120/70 mm Hg. The primary end point was the change from baseline in the mean 24-hour ambulatory systolic BP after 12 weeks of treatment. The baseline mean ± SD seated cuff systolic/diastolic BP was 158.8 ± 13.1/89.1 ± 10.1 mm Hg and the mean ± SD 24-hour ambulatory systolic/diastolic BP was 144.4 ± 11.7/81.6 ± 9.8 mm Hg. At week 12, the change from baseline in the mean ± SEM 24-hour ambulatory systolic/diastolic BP was −19.9 ± 0.8/−11.2 ± 0.5 mm Hg (p <0.0001 vs baseline), and 70% of patients had achieved a 24-hour ambulatory BP target of <130/80 mm Hg. At the end of 18 weeks of active treatment in patients uptitrated to amlodipine/OM 10/40 mg plus hydrochlorothiazide 25 mg, the change from baseline in the mean ± SEM seated BP was −28.0 ± 1.5/−13.7 ± 1.0 mm Hg (p <0.0001 vs baseline), with 62% of patients reaching the guideline-recommended seated BP goal of <130/80 mm Hg. Drug-related treatment-emergent adverse events occurred in 19.3% of patients. The most frequent events were peripheral edema (6%), dizziness (3%), and hypotension (2%). In conclusion, this amlodipine/OM-based titration regimen was well tolerated and effectively lowered BP throughout the 24-hour dosing interval in patients with hypertension and type 2 diabetes.
The AZOR Trial Evaluating Blood Pressure Reductions and Control (AZTEC) study showed that an amlodipine/olmesartan medoxomil (OM)-based titration regimen controlled blood pressure (BP) for a 24-hour period in patients with hypertension. The present study is a Prospective, open-label, ambulatory BP monitoring study to evaluate the safety and Efficacy of an olmesartan medoXomil- and amlodipine-based treatment regimen in patients with type 2 diabetes and hypertension (APEX). The efficacy and tolerability were evaluated after 12 weeks of treatment. In addition, the effect of adding hydrochlorothiazide (HCTZ) to the regimen for an additional 6 weeks was assessed.
Methods
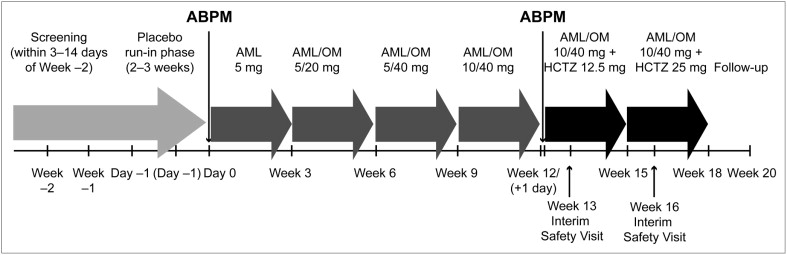
The present phase IV, open-label, multicenter, single-arm, titration study consisted of a 3- to 14-day screening period, a 2- to 3-week placebo run-in period, and an 18-week active treatment phase ( Figure 1 ). At each clinic visit, the seated BP and heart rate were determined in triplicate from the patient’s nondominant arm using an automated Omron device. A Spacelabs Healthcare 90207 Oscillometric device (Issaquah, WA) was used to perform 24-hour ambulatory BP monitoring at baseline and after 12 weeks of active treatment. The monitoring was started at 8 a.m. ± 2 hours immediately before dosing. The same time window was monitored at baseline and week 12. The present study was conducted in accordance with the institutional review boards’ committee regulations and the Declaration of Helsinki. The institutional review board oversaw the study at each site. All patients provided informed consent before study participation.
Noninstitutionalized men or women aged 18 to 80 years with type 2 diabetes mellitus were eligible for the present study if they were undergoing stable treatment with any oral antidiabetic agent for ≥3 months. The patients not receiving oral antidiabetic agents were required to have a fasting plasma glucose level of ≥126 mg/dl (7.0 mmol/L) at screening and a documented history of type 2 diabetes according to the American Diabetes Association criteria (i.e., fasting plasma glucose ≥126 mg/dl [7.0 mmol/L]; hyperglycemia symptoms, and a casual plasma glucose level ≥200 mg/dl [11.1 mmol/L]; or 2-hour plasma glucose level ≥200 mg/dl [11.1 mmol/L] during an oral glucose tolerance test). Patients with newly diagnosed or uncontrolled hypertension (i.e., current antihypertensive medication) were identified at screening if the mean systolic BP was >130 mm Hg and/or the diastolic BP was >80 mm Hg. The patients entered the active treatment phase if they had taken ≥80% of the study medication and had a mean seated systolic BP of ≥140 and ≤199 mm Hg and a mean seated diastolic BP of ≤114 mm Hg at 2 consecutive study visits during the placebo run-in period, with a difference in the seated systolic BP of ≤15 mm Hg between the 2 qualifying study visits. Additional inclusion criteria determined by ambulatory BP monitoring were a mean daytime (8 a.m. to 4 p.m. ) systolic BP of ≥130 and ≤199 mm Hg and diastolic BP of ≤114 mm Hg after the placebo run-in.
Patients with uncontrolled hypertension with multiple antihypertensive therapies, type 1 or 2 diabetes requiring insulin, type 2 diabetes with a glycosylated hemoglobin level of ≥9.0%, proteinuria of >1+ on the dipstick, serum creatinine >1.7 mg/dl, or fasting serum glucose level of >300 mg/dl at screening were excluded. A nondominant arm circumference of <24 or >42 cm or serious disorders that could have limited the ability to evaluate the efficacy or safety of OM (e.g., cardiovascular and renal disease) were also exclusionary. Women were eligible only if they were not pregnant or planning to become pregnant, were not breastfeeding, and were practicing protocol-approved birth control. The use of antihypertensive agents other than OM, amlodipine, or HCTZ was prohibited.
All patients started active treatment with amlodipine 5 mg/day at approximately 8.00 a.m. The medication was uptitrated at 3-week intervals if the patients’ mean seated BP was greater than the titration threshold of ≥120/70 mm Hg. The titration schedule was amlodipine/OM 5/20, 5/40, and 10/40 mg/day, followed by the addition of HCTZ 12.5 and 25 mg/day. The patients with a mean seated systolic BP of ≥130 mm Hg were immediately uptitrated to HCTZ 12.5 and 25 mg/day. An amendment was added to the study design for patients who required uptitration to HCTZ after week 12. The patients with a mean seated systolic BP of ≥120 and <130 mm Hg underwent orthostatic BP and heart rate assessments before the initiation or uptitration of HCTZ. If these patients had an orthostatic systolic BP reduction of >10 mm Hg but were not taking HCTZ, they maintained their current therapy (amlodipine/OM 10/40 mg). Patients with an orthostatic systolic BP reduction of ≤10 mm Hg received HCTZ but underwent an additional safety assessment 1 week later. Patients receiving HCTZ 12.5 or 25 mg with an orthostatic decrease in systolic BP of >10 mm Hg at these interim safety assessments were withdrawn from the present study. Throughout the active treatment period, the patients with a mean seated BP of <120/70 mm Hg at any visit maintained their current therapy but were uptitrated to the next dose level if their seated systolic BP had increased to ≥130 mm Hg and/or their seated diastolic BP had increased to ≥80 mm Hg at any subsequent visit. The patients with a mean seated systolic BP of ≥200 mm Hg or seated diastolic BP ≥115 mm Hg or seated systolic BP of <120 mm Hg or seated diastolic BP of <70 mm Hg with symptomatic hypotension at any visit were required to withdraw from the study.
The primary efficacy variable was the change from baseline in the mean 24-hour ambulatory systolic BP after 12 weeks of treatment. The secondary ambulatory BP efficacy variables included the change from baseline in the mean 24-hour ambulatory diastolic BP, the change from baseline in the mean daytime (8.00 a.m. to 4.00 p.m. ) and nighttime (12.00 a.m. to 6.00 a.m. ) ambulatory BP, and the change from baseline in the mean ambulatory BP during the last 6, 4, and 2 hours of the 24-hour dosing interval after 12 weeks of treatment. These variables also included the proportions of patients achieving the prespecified ambulatory BP targets for these same periods and the proportion of patients who were “nondippers” at baseline and converted to “dippers” after 12 weeks of treatment. Ambulatory BP “dippers” were defined as patients with a nocturnal ambulatory systolic BP and/or diastolic BP reduction of ≥10% of their mean daytime ambulatory systolic and/or diastolic BP. Other secondary efficacy variables included changes from baseline in the mean seated BP at each titration step up to week 18, the proportion of patients achieving the prespecified ambulatory and seated systolic and diastolic BP reductions, and the proportion of patients achieving the seated BP goals during the study.
The adverse events reported by patients or observed by the investigators at any visit during the 18-week active treatment period and through 14 days after the last dose was taken were recorded and graded as mild, moderate, or severe and unrelated, unlikely related or possibly, probably, or definitely treatment-related. Serious adverse events were any events that resulted in death, were life-threatening, required inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability/incapacity, or were an important medical event. Safety was also assessed by monitoring the laboratory values and physical examination findings at screening, week 12, and week 18.
The treated group included all subjects who had received one or more doses of active study medication. All efficacy (except ambulatory BP monitoring analysis) and safety analyses were based on the treated group. All ambulatory BP monitoring analysis was based on the ambulatory BP monitoring group, which consisted of all treated subjects having both valid baseline and week 12 ambulatory BP monitoring data. Summary statistics were calculated for the continuous variables, and the numbers and percentages of patients were calculated for the categorical variables. For the number of patients achieving certain BP goals by titration dose, the cumulative achievement rate (the percentage of patients achieving their goal at any point on or before the specified dose was taken) was calculated. The primary efficacy variable was analyzed using a one-sample t test, and the mean, 2-sided 95% confidence interval, and standard error of the mean were provided. The statistical test had a 2-sided significance level of 5%. It was estimated that a sample size of 200 patients would provide 99% power for a significant change from baseline in mean 24-hour ambulatory systolic BP at 12 weeks. This sample size was expected to allow approximately 150 patients to complete the first 12 weeks of the active treatment period.
Results
The study was conducted at 33 centers in the United States. The patient disposition is presented in Figure 2 . All 207 patients who entered the active treatment phase received one or more doses of the study medication, and 164 (79%) completed the study. The main reasons for study withdrawal were adverse events (n = 12), patient request (n = 10), protocol violation (n = 10), and lost to follow-up (n = 7).
The baseline characteristics of the treated group (n = 207) are listed in Table 1 . Before the placebo run-in, 183 patients of the treated group were undergoing antihypertensive therapy. The mean study drug exposure was 112.9 days (range 1 to 158). At week 18, 7 patients were receiving amlodipine/OM 5/20 mg, 5 amlodipine/OM 5/40 mg, 14 amlodipine/OM 10/40 mg, 38 amlodipine/OM 10/40 mg plus HCTZ 12.5 mg, and 101 amlodipine/OM 10/40 mg plus HCTZ 25 mg.
| Characteristic | Treated Group (n = 207) |
|---|---|
| Age at screening (years) | 59.1 ± 9.5 |
| Age <65 years | 145 (70%) |
| Gender | |
| Men | 122 (59%) |
| Women | 85 (41%) |
| Race | |
| Caucasian | 163 (79%) |
| Black | 35 (17%) |
| Asian | 8 (4%) |
| Other | 2 (1%) |
| Hispanic or Latino ethnicity | 54 (26%) |
| Weight (kg) | 94.3 ± 20.2 |
| Hemoglobin A1c (%) (n = 206) | 6.9 ± 0.8 |
| Body mass index (kg/m 2 ) | 32.8 ± 5.9 |
| Obesity | 49 (24%) |
| Dyslipidemia | 31 (15%) |
| Hypercholesterolemia | 61 (30%) |
| Hyperlipidemia | 50 (24%) |
| Mean interval from diabetes diagnosis to screening (years) | 6.4 |
| Seated BP (mm Hg) | 158.8 ± 13.1/89.1 ± 10.1 |
| Baseline cuff BP (mm Hg) | |
| 140–159/90–99 | 115 (56%) |
| ≥160/≥100 | 92 (44%) |
| 24-Hour ambulatory BP (mm Hg) | 144.4 ± 11.7/81.6 ± 9.8 |
The baseline and week 12 data for 24-hour ambulatory BP were available for 165 patients. The mean 24-hour ambulatory BP was 144.3/81.6 mm Hg at baseline and 124.3/70.4 mm Hg at week 12 ( Figure 3 ). At week 12, the overall change from baseline in the mean 24-hour ambulatory systolic BP (primary efficacy variable) was −19.9 mm Hg (p <0.0001; Figure 4 ). The reduction in the mean ± SEM 24-hour ambulatory diastolic BP was −11.2 ± 0.5 mm Hg (p <0.0001; Figure 4 ). The ambulatory BP was also significantly reduced in the daytime and nighttime and during the last 6, 4, and 2 hours of the dosing interval at week 12 (all p <0.0001 vs baseline; Figure 4 ). After 12 weeks of treatment, 70%, 46%, and 36% of patients had achieved the prespecified 24-hour ambulatory BP targets of <130/80, <125/75, and <120/80 mm Hg, respectively. During the daytime, 50%, 29%, and 26% of the patients had achieved these thresholds compared to 83%, 72%, and 68% of the patients during the nighttime.
The mean seated BP at baseline was 158.8/89.1 mm Hg and was, in general, progressively reduced during each titration step ( Figure 5 ). All seated BP reductions from baseline were statistically significant during each dosing period (p <0.0001). The change from baseline in the mean seated BP ± SEM at the end of each titration period for the patients receiving monotherapy (amlodipine), dual therapy (amlodipine/OM), and triple therapy (amlodipine/OM plus HCTZ) was −10.4 ± 0.9/−4.1 ± 0.5 mm Hg, −23.0 ± 1.0/−10.8 ± 0.6 mm Hg, and −30.1 ± 1.3/−14.9 ± 0.8 mm Hg, respectively (all p <0.0001 vs baseline). Intensifying treatment from amlodipine monotherapy to amlodipine/OM dual therapy produced a reduction in the mean seated BP ± SEM of 12.5 ± 1.0/6.7 ± 0.6 mm Hg. The observed titration effect of adding HCTZ to amlodipine/OM was 8.5 ± 1.0/4.8 ± 0.6 mm Hg. The cumulative proportions of patients achieving a seated BP target of <140/90 mm Hg, a goal of <130/80 mm Hg, and a target of <120/80 mm Hg at any time point by titration period is presented in Figure 6 . The amlodipine/OM plus HCTZ-based titration regimen facilitated the cumulative achievement of the recommended seated BP goal of <130/80 mm Hg at any time point in 62% of the patients.



