Chapter 10
Management of Acute Coronary Syndrome
Introduction and Epidemiology of Acute Coronary Syndrome
Cardiovascular disease is the most common cause of mortality. Each year it accounts for 30% of deaths worldwide and 38.5% in the United States and Western Europe [1]. Although the incidence of cardiovascular disease is decreasing in high-income countries due to education and advancement in medical therapy, it is rapidly rising in middle- and low-income countries as they become increasingly industrialized and urbanized [1]. Acute coronary syndrome (ACS) is a spectrum of unstable cardiovascular disease that includes unstable angina (UA), non-ST-elevation myocardial infarction (NSTEMI), and ST-elevation myocardial infarction (STEMI), and is often the initial clinical presentation for coronary artery disease (CAD). The major risk factors for ACS include tobacco abuse, family history of CAD, advanced age, hypertension, hyperlipidemia, and diabetes mellitus.
This chapter focuses on the management of ACS in patients with diabetes. Diabetics are more likely than nondiabetics to experience ACS, and diabetes is an independent predictor for mortality in ACS. Diabetics are also more likely to develop complications of ACS and its management such as heart failure and bleeding. With a few exceptions, the management of ACS is similar in patients with and without diabetes. In patients with diabetes, management does not differ between patients who are insulin dependent and patients who do not require insulin. Although intensive treatment of hyperglycemia, dyslipidemia, and hypertension can reduce cardiovascular and microvascular events in diabetics by as much as 50%, diabetes remains a major risk factor for ACS [2].
Pathophysiology
The initiating event in ACS is rupture or erosion of an atherosclerotic plaque within the coronary endothelium. This exposes the lumen of the coronary artery to subendothelial matrix, leading to the activation of platelets and eventual thrombus formation. ACS is a dynamic process, during which there is cyclical transition among partial vessel occlusion, complete occlusion, and reperfusion. In UA, plaque rupture results in severe obstruction of coronary blood flow and subsequent ischemic symptoms, at times associated with electrocardiographic ST depressions or T-wave inversions without elevation of cardiac biomarkers. NSTEMI is defined by obstruction that leads to infarction without electrocardiographic ST elevation. Plaque rupture that results in complete and prolonged coronary artery occlusion usually leads to ST elevation and subsequent myocardial infarction.
Clinical Presentation
Classically, patients with ACS present with substernal chest pain characterized as “vice-like” or “pressure.” The pain often radiates to the left shoulder or jaw, but can radiate to the right arm, back, neck, and/or epigastrium. Associated symptoms include nausea, vomiting, diaphoresis, palpitations, dyspnea, dizziness, and/or confusion. The chest discomfort associated with STEMI is usually more severe than the discomfort experienced with UA/NSTEMI. Also, chest pain in the setting of STEMI is less likely to be relieved by nitroglycerine. Atypical symptoms or silent ischemia (with associated symptoms such as cardiac dysrhythmias or heart failure) are more common in diabetics, women, and the elderly. Appropriate therapy for ACS is dependent on early recognition of symptoms by the patient, as delayed medical evaluation can result in cardiogenic shock and sudden cardiac death. While physical examination is unlikely to aid in the diagnosis of ACS, it is important in risk stratification, excluding alternate diagnoses, and determining whether mechanical complications of myocardial infarction (MI) are present.
Differential Diagnosis
Other diagnoses that cause chest pain can be mistaken for ACS. Gastrointestinal disorders such as gastroesophageal reflux disease (GERD), esophageal spasm, and esophageal hyperalgesia can mimic ischemic chest pain. GERD and CAD can coexist, and it is not uncommon for patients with ACS to mistake their symptoms as reflux. Given this, it is prudent to exclude ACS before proceeding with further evaluation for gastrointestinal disease.
Acute pericarditis causes pleuritic chest pain that is worse when supine and relieved by sitting up and leaning forward. It is usually associated with diffuse ST-segment elevation and concomitant PR-segment depression. If there is myocardial involvement (myocarditis), the cardiac biomarkers may be elevated and transthoracic echocardiogram (TTE) may reveal a regional wall motion abnormality. It can be distinguished from acute MI by the lack of reciprocal ST depression. Also, the ST elevations in acute pericarditis are usually concave, as opposed to the convex ST elevations seen in acute MI.
Aortic dissection typically causes sharp, tearing chest pain with radiation to the back. It is often most severe at onset, as opposed to ischemic chest pain, the severity of which increases over time. Aortic dissection is usually distinguished from ACS based on symptoms, though physical examination can assist in the diagnosis. Examination in patients with aortic dissection may reveal a difference between right and left upper extremity blood flow, detected by comparing the pulse and blood pressure. The diastolic murmur of aortic insufficiency may also be present. If the aorta is enlarged, the chest radiograph will show a widened mediastinum. A TTE may reveal the dissection flap, but the diagnosis is usually made by transesophageal echocardiogram (TEE), computerized tomography (CT), or magnetic resonance imaging (MRI).
Acute onset of pleuritic chest pain and shortness of breath in the absence of pathology on chest radiography suggests pulmonary embolism (PE). The ECG most often shows sinus tachycardia, but may demonstrate right ventricular strain. Although cardiac biomarkers can be slightly elevated with acute PE, TTE can be performed to rule out a left ventricular wall motion abnormality and identify right ventricular dysfunction.
Diagnosis
The diagnosis of ACS should be suspected in any patient with chest pain and risk factors for CAD. The most important initial investigation is a 12-lead electrocardiogram (ECG). This will help distinguish STEMI from UA or NSTEMI. If the ECG reveals ST-segment elevation in a pattern that would suggest MI, the focus of care should shift to emergent myocardial reperfusion. By definition, there should be 1-mm ST-segment elevation in two or more contiguous leads. In acute MI, the ST-segment elevations are usually convex. It is important to be aware of other diagnoses that may cause ST-segment elevation without concomitant ischemia. These diagnoses are listed in Table 10.1.
Table 10.1 Nonischemic causes of electrocardiographic ST-segment elevation. (Source: Adapted from Wang et al. [3].)
| Early repolarization |
| Left ventricular hypertrophy |
| Left bundle branch block |
| Male pattern |
| Hyperkalemia |
| Acute pericarditis |
| Brugada syndrome |
| Pulmonary embolism |
| Postcardioversion |
There are a few situations in which the patient presents with an acute MI but the ECG does not reveal classic ST-segment elevation. The first is posterior MI due to circumflex coronary artery occlusion, which because of its location causes anterior ST-segment depression and tall R waves. It is also possible for a posterior MI to be electrically silent, and thus patients with an occluded left circumflex artery can have a normal ECG [4]. Another situation in which classic ST-segment elevation may be absent is left bundle branch block (LBBB). LBBB “not known to be chronic” in the appropriate clinical setting is considered a STEMI equivalent for two reasons. First, in the setting of proximal left anterior descending (LAD) coronary artery occlusion, new LBBB can occur because of lack of blood flow to the left bundle through the first septal perforator. Next, an existing LBBB due to conduction disease can impair the diagnosis of subsequent MI. If the diagnosis of MI is in question, a TTE can assist the clinician by identifying a regional wall motion abnormality. Once the diagnosis of STEMI is made, eligible patients should receive emergent reperfusion without waiting for results of cardiac biomarker testing. Post-reperfusion cardiac biomarkers can aid in determining the size of myocardial infarction.
By definition, the ECG in UA and NSTEMI does not show ST-segment elevation. Rather, the ECG may be normal or show ST-segment depression and/or T-wave inversion. UA and NSTEMI cannot be differentiated based on ECG changes, but instead by cardiac biomarker analysis. In UA cardiac biomarkers are within normal limits, whereas with NSTEMI these markers are elevated. After the onset of MI, it can take up to four hours for these biomarkers to be released into the blood stream. Thus, a patient who presents with MI two hours after the onset of chest pain may have initially normal cardiac biomarkers. In patients with UA and NSTEMI, it is therefore important to check at least two sets of biomarkers (preferably three) drawn a minimum of four hours apart. The most commonly used cardiac biomarkers and their timing of release are shown in Figure 10.1. Troponin T and I are highly sensitive and available at most healthcare facilities. A troponin T measured 72 hours after acute MI may predict infarct size [5, 6], 12. Because of its high sensitivity, troponin elevation can occur in the setting of other conditions such as PE and congestive heart failure (CHF). Creatinine kinase (CK) and creatinine kinase myocardial band (CK-MB) are also elevated in myocardial infarction. Like troponin, CK is helpful in determining the size of myocardial infarction. CK is also important for determining reinfarction as it normalizes 24 hours after MI, unlike CK-MB and troponin, which may remain elevated for several days.
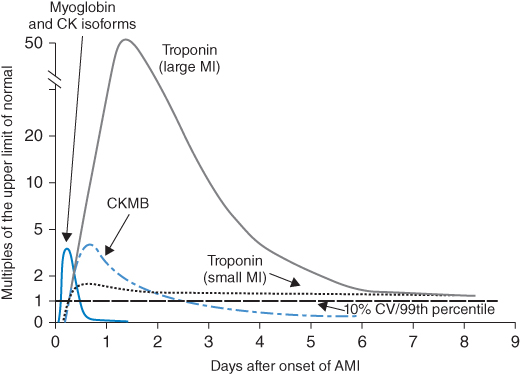
Figure 10.1 Timing of cardiac biomarker release after myocardial infarction. (Source: Anderson et al. 2007 [5]. Reproduced with permission of Elsevier.)
Risk Stratification
Estimating risk of death is useful in patients with ACS, as it can aid in treatment decisions and in the counseling of patients and their families. Risk models have been developed to assist the clinician in risk prediction. The Global Registry of Acute Coronary Events (GRACE) score predicts in-hospital mortality in all patients with ACS [9]. Calculation involves multiple variables, including age, heart rate, Killip class (Table 10.2), systolic blood pressure, creatinine level, presence or absence of cardiac arrest on admission, presence or absence of cardiac biomarkers, and ST-segment changes. A score of 250 or greater predicts a >50% chance of in-hospital death.
Table 10.2 Killip class and estimated 30-day mortality. (Source: Adapted from Lee et al. [8, 7]. Reproduced with permission of Wolters Kluwer Health).
| Killip class | Characteristics | Mortality rate (%) |
| I | No signs of heart failure | 5.1 |
| II | Rales, JVD, S3 gallop | 13.6 |
| III | Pulmonary edema | 32.2 |
| IV | Cardiogenic shock | 57.8 |
JVD, jugular venous distention.
In STEMI, the most important risk factor for 30-day mortality is age, followed by systolic blood pressure, Killip classification, heart rate, and location of MI (Table 10.3). The thrombolysis in myocardial infarction (TIMI) risk model (Table 10.4) for patients with STEMI incorporates these and other variables obtained from the history, physical exam, and ECG. A TIMI risk score of 9 or greater predicts a 30-day mortality of 35%. By comparison, 30-day mortality is <2% in STEMI patients with a score of 0 to 1.
Table 10.3 Location of myocardial infarction and mortality. (Source: Adapted from Topol 1998 [6]. Reproduced with permission of Lippincott Williams & Wilkins.)
| Location | 30-day mortality rate (%) | 1-year mortality rate (%) |
| Proximal LAD | 19.6 | 25.6 |
| Mid LAD | 9.2 | 12.4 |
| Distal LAD | 6.8 | 10.2 |
| Proximal RCA or LCx | 6.4 | 8.4 |
| Distal RCA or LCx | 4.5 | 6.7 |
LAD, Left anterior descending; LCx, Left circumflex; RCA, Right coronary artery.
Table 10.4 TIMI risk model for prediction of 30-day mortality in patients with ST-segment elevation myocardial infarction. (Source: Adapted from Morrow 2000 [9]. Reproduced with permission of Wolters Kluwer Health.)
| History | Points |
| 65–74 years old | 2 |
| ≥75 years old | 3 |
| Angina or DM/HTN | 1 |
| Physical Exam | |
| HR >100 bpm | 2 |
| SBP <100 mmHg | 3 |
| Killip class II–IV | 2 |
| Weight <67 kg | 1 |
| Presentation | |
| Time to treatment >4 hrs | 1 |
| Anterior ST elevation or LBBB | 1 |
| TIMI risk score = (0–14) points | |
DM, diabetes mellitus; HR, heart rate; HTN, hypertension; SBP, systolic blood pressure.
There is also a TIMI risk score for patients with UA and NSTEMI (Table 10.5). The score consists of seven variables extracted from the patient’s history, ECG, and cardiac biomarker analysis. A score of 6 or greater predicts a 41% incidence of all-cause mortality, MI, or severe recurrent ischemia requiring revascularization. The ischemic complication event rate with a score of 0 to 1 is 4.7%. Not only can this risk model be used to predict outcomes in patients with UA/NSTEMI, but it can also serve as a clinical decision-making tool for determining which patients should receive early coronary angiography. In the TACTICS-TIMI 18 (Treat Angina with Aggrastat and Determine Cost of Therapy with an Invasive or Conservative Strategy-Thrombolysis in Acute Myocardial Infarction) trial, a TIMI risk score of 3 or greater favored an early (within 48 hours) invasive strategy, whereas patients with a score of 2 or less had better outcomes with conservative therapy [13].
Table 10.5 TIMI risk model for predicting 14-day outcomes in patients with UA/NSTEMI. (Source: Data from Antman [10].)
| Characteristics | Points |
| ≥1 mm ST deviation on ECG | 1 |
| ≥2 episodes of angina in the preceding 24 hours | 1 |
| ≥3 risk factors for CAD | 1 |
| Elevated cardiac biomarkers | 1 |
| ≥50% stenosis on prior left heart catheterization | 1 |
| ≥65 years of age | 1 |
| Use of aspirin in the past 7 days | 1 |
| TIMI risk score = (0–7) points | |
Management
With the exception of reperfusion timing, patients across the spectrum of ACS are managed similarly. The major difference is that STEMI patients need emergent reperfusion, whereas patients with UA and NSTEMI can be risk stratified to early coronary angiography or conservative management. The following discussion will include medical management and reperfusion strategies in patients with ACS. The management of patients with and without diabetes is similar, with a few exceptions that will be discussed below.
Initial Medical Therapy
Anti-ischemic Therapy
The three classes of drugs used to reduce myocardial ischemia in patients presenting with ACS are beta-blockers, nitrates, and calcium channel blockers. Beta-blockers reduce myocardial oxygen demand by reducing blood pressure, heart rate, and contractility [11]. Although the benefit of long-term beta-blocker therapy in ACS patients has been well established by clinical trials, there are no randomized data in the era of percutaneous coronary intervention (PCI) to suggest that early administration of beta-blocker therapy reduces mortality [14, 15]. The COMMIT/CCS-2 (Clopidogrel and Metoprolol in Myocardial Infarction Trial/Second Chinese Cardiac Study) trial randomized 45,852 patients with acute MI to intravenous metoprolol followed by oral administration until discharge, or, if prolonged hospitalization, a maximum of four weeks [14]. While there was a reduction in reinfarction and ventricular fibrillation (VF), there was no improvement in mortality, mainly due to an increase in cardiogenic shock [14]. Based on these results, current European Society of Cardiology (ESC) and American Heart Association (AHA) guidelines caution against early administration of beta-blocker therapy in STEMI patients at risk for developing cardiogenic shock [15, 16]. In patients with UA and NSTEMI, in-hospital administration of these agents may prevent progression of ACS and reduce mortality [11]. For instance, the CRUSADE (Can Rapid Risk Stratification of Unstable Angina Patients Suppress Adverse Outcomes with Early Implementation of the ACC/AHA Guidelines) registry demonstrated that patients with UA and NSTEMI who received beta-blockade had a 34% reduction in in-hospital mortality (3.9% vs. 6.9%, p < 0.001) [17]. Because of confounding that exists in nonrandomized analyses, these data should be interpreted carefully. Similar to STEMI patients, guidelines caution against aggressive early administration of these agents in UA/NSTEMI patients at risk for developing cardiogenic shock.
Nitrates reduce myocardial oxygen demand through venodilatation, which reduces myocardial preload and thus ventricular wall stress [11, 18]. Nitroglycerine also causes vasodilatation of normal and atherosclerotic coronary arteries, thus improving myocardial blood flow [11, 18]. Although no randomized, placebo-controlled trials have been performed to assess the efficacy of nitroglycerine in improving symptoms or reducing cardiac events, these agents are commonly administered on the basis of observational data. Nitrates are particularly useful in the setting of persistent chest pain, hypertension, or congestive heart failure (CHF). Nitroglycerine can be administered orally, topically, or intravenously. Intravenous nitroglycerine is preferred in patients with persistent ischemic symptoms, because it is easily titratable. The dose is 10 to 20 mcg/min with 5–10 mcg/min increases every 5–10 minutes. Nitrates should be used with caution in STEMI patients suspected of having right ventricular infarction, as these patients are preload dependent. Patients taking phosphodiesterase-5 inhibitors (sildenafil, vardenafil, tadalefil) should not receive nitroglycerine, as this combination of therapy may result in profound hypotension [11, 18].
Calcium channel blockers improve myocardial ischemia by reducing vascular smooth muscle contractility, which results in coronary artery vasodilatation. These agents are not first line for the treatment of ischemia, but are rather given to patients with persistent symptoms despite therapy with beta-blockers and nitrates [11, 18]. As they are contraindicated in the setting of CHF or impaired left ventricular function, calcium channel blockers are rarely used in patients with STEMI. In patients with UA and NSTEMI, diltiazem and verapamil are the preferred agents, as their efficacy in reducing myocardial ischemia is similar compared to beta-blockers [19, 20]. Because of reflex sympathetic activation, nifedipine and other dihydropiridine calcium channel blockers should be avoided in patients with ACS unless combined with a beta-blocker [11, 18, 21].
Antiplatelet Therapy
According to both ESC and AHA guidelines, all patients with ACS should receive dual antiplatelet therapy (DAPT) [11, 16–18, 22, 23]. One of the antiplatelet agents should be aspirin at an oral dose of 150–325 mg. Nonenteric coated aspirin is preferred for more rapid absorption [11]. Current options for the second antiplatelet agent include an oral platelet P2Y12 receptor antagonist or an intravenous platelet glycoprotein IIb/IIIa receptor inhibitor (GPI). There are three P2Y12 receptor antagonists currently available for use in ACS. Clopidogrel and prasugrel are thienopyridines that irreversibly bind the platelet P2Y12 receptor, and ticagrelor is a triazolopyrimidine that reversibly inhibits the same platelet receptor [11, 16–18, 22].
Clopidogrel is a pro-drug that is poorly metabolized to its active metabolite. Because of this, it is the least potent of the P2Y12 receptor antagonists. The benefit of clopidogrel in ACS has been extensively studied in randomized trials. In the large, randomized CURE (Clopidogrel in Unstable Angina to Prevent Recurrent Events) trial, patients who received clopidogrel had a significant reduction in the primary composite endpoint of cardiovascular death, MI, and stroke (9.3% vs. 11.4%, p < 0.001). The benefit persists in patients who receive PCI. A subset analysis, PCI-CURE, compared outcomes in the 2,658 patients who received PCI and found a significant reduction in cardiovascular death, MI, and urgent target vessel revascularization in the patients treated with clopidogrel (4.5% vs. 6.4%, p = 0.03) [24]. Clopidogrel is given as a loading dose of 300–600 mg, and continued at a dose of 75 mg daily. The 600 mg loading dose has improved onset of action and platelet inhibition, and in patients undergoing PCI is associated with improved ischemic outcomes compared to 300 mg [11, 16–18, 22, 23]. The CURRENT-OASIS 7 (Committee members of the Clopidogrel and Aspirin Optimal Dose Usage to Reduce Recurrent Events – Seventh Organization to Assess Strategies in Ischemic Syndromes) trial compared a 300 mg clopidogrel loading dose to a 600 mg loading dose in patients with ACS. Although the higher loading dose did not reduce the primary composite endpoint of cardiovascular death, MI, or stroke in the overall study population, it did significantly reduce the primary endpoint in patients who received PCI (3.9% vs. 4.5%, p = 0.04) [25]. Clopidogrel should be discontinued five days prior to major surgery [11].
Prasugrel is a pro-drug with more complete and rapid metabolism to active drug, and thus is superior to clopidogrel in onset of action and strength of platelet inhibition. It is given as a loading dose of 60 mg and maintained at a dose of 10 mg daily. Prasugrel was compared to clopidogrel in the TRITON-TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel–Thrombolysis In Myocardial Infarction) trial, which randomized ACS patients to either 300 mg of clopidogrel or 60 mg of prasugrel for up to 15 months [26]. The primary outcome of cardiovascular death, MI, or stroke was reduced in patients who received prasugrel (9.9% vs. 12.1%, p < 0.001), which was driven mainly by a reduction in MI (7.3% vs. 9.5%, p < 0.001) [26]. Patients who received prasugrel had an increase in major bleeding, particularly patients greater than 75 years of age or with body weight less than 60 kg. In addition, post hoc analysis showed worse outcomes in patients with a history of transient ischemic attack (TIA) or stroke, and thus prasugrel should be avoided in these patients [26]. A subgroup analysis showed a substantially greater treatment effect of prasugrel compared to clopidogrel in diabetics compared to nondiabetics (primary outcome in diabetics: 12.2% vs. 17%, HR 0.70, p < 0.001; in nondiabetics: 9.2% vs. 10.6%, HR 0.80, p = 0.02). These results should be interpreted carefully, as they are subject to the same potential for spurious findings as with any subgroup analysis. Prasugrel should be discontinued seven days before major surgery [11].
Ticagrelor is the newest of the platelet P2Y12 inhibitors. It is given as a loading dose of 180 mg and continued at a dose of 90 mg twice daily. Because it is administered in active form, the onset of action is rapid (30 minutes) and the platelet inhibition is consistent [11]. Ticagrelor is the only P2Y12 inhibitor that has been shown in a randomized trial to provide mortality benefit in ACS [27]. The PLATO (Platelet Inhibition and Patient Outcomes) trial compared ticagrelor to clopidogrel for 12 months in 18,624 patients presenting with all forms of ACS [27]. The primary composite endpoint of death from vascular causes, MI, and stroke was significantly reduced in patients who received ticagrelor (9.8% vs. 11.7%, p < 0.001). The individual endpoints of death from vascular causes (4.0% vs. 5.1%, p = 0.001) and death from any cause (4.5% vs. 5.9%, p
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


