Lymphoproliferative Disorders |
Lymphoproliferative disorders of the lung and pleura comprise a varied but rare group of localized and diffuse processes that span the morphologic gamut from reactive to neoplastic and include several peculiar lesions that do not fit conventional definitions of either hyperplasia or neoplasia. Although most diagnoses are based on light microscopy, immunohistochemical and molecular investigations have assumed a vital role. Nomenclature and classification schemes have undergone drastic changes over the past quarter century and current definitions appear reasonable. Malignant lesions are best classified according to the current World Health Organization (WHO) scheme.1 This chapter presents the clinicopathologic features of primary and secondary pulmonary and pleural hematolymphoid lesions.
ANATOMY AND HISTOLOGY OF THE PULMONARY LYMPHOID SYSTEM
Pulmonary lymphatics are divided into two interconnecting channels that drain to peribronchial, hilar, and/or mediastinal lymph nodes and eventually into the thoracic duct, right lymphatic duct, and subclavian veins.2 One system drains through the visceral pleura around the lung into mediastinal lymph nodes and the other drains from central lung parenchyma to peribronchial and hilar lymph nodes. The lymphatics communicate at lobar, lobular, and pleural boundaries and thus serve each other as potential collaterals.3,4 Although not usually obvious in histologic sections of normal lung, lymphatics are prominent in disease states ranging from pulmonary edema to lymphangitic carcinoma. In the latter, lymphatic channels distended with malignant cells are apparent within the visceral pleura, interlobular septa, and adventitia of arteries, veins, and bronchioles. Of note, alveolar septa do not contain lymphatic channels. All lymphatics contain valves and flat endothelial cells line the discontinuous basal lamina. Larger lymphatics contain smooth muscle and collagen.
Small submucosal aggregates of lymphoid cells are often prominent at bronchial bifurcations and near distal respiratory bronchioles (pulmonary microtonsils) and represent bronchus-associated lymphoid tissue (BALT) (Fig. 120-1).5–7 Whether humans are born with this specialized secondary lymphoid system, or whether the aggregates of B lymphocytes, T lymphocytes, HLA-DR+ interdigitating cells, follicular dendritic cells, and lymphoid follicles with overlying flattened and attenuated specialized epithelium develop in response to antigenic stimulation is controversial.8,9 Viruses, connective tissue disorders, tobacco use, and obstructive pneumonia are just a few pathologic processes known to induce BALT (Table 120-1). Unlike typical lymph nodes that rely on afferent lymphatics for antigen retrieval, BALT is integrated into lung tissue and antigen is sampled directly from the bronchial and bronchiolar lumens through specialized “lymphoepithelium.” Immunoglobulins, most notably IgA, are synthesized and secreted by lymphocytes directly into airway lumens. Amazingly, this system appears capable of mounting a competent adaptive immune response. In addition, BALT B-lymphocytes circulate and “home” to other mucosal sites such as the conjunctiva, salivary glands, stomach, and intestines to create a common mucosal immune system, the mucosa-associated lymphoid system (MALT).6 Thus, responses induced in one location can be replicated at other sites.10 Malignant lymphoma arising in one MALT location can secondarily involve other MALT sites. BALT appears to be the origin of many primary pulmonary lymphoid lesions.11
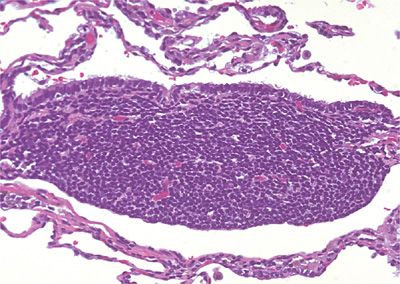
Figure 120-1 Bronchus-associated lymphoid tissue. The submucosal collection of lymphoid cells is intimately associated with overlying bronchiolar epithelium. Hematoxylin and eosin, 40× original magnification.
Intrapulmonary lymph nodes (IPLs) may be part of the pulmonary immune system and may also be induced by antigenic stimuli rather than normal embryologic development.12 Autopsy studies suggest a prevalence of 18%, and although many are related to bronchi of the first few orders, peripheral subpleural locations are not uncommon. In this age of high-resolution computed tomography (HRCT) and lung cancer screening programs, up to 80% of reported cases of IPLs occur in men with histories of tobacco use and almost 35% of cases are multiple.13 They appear as round to angulated sharply circumscribed subpleural opacities up to 2 cm in diameter and are found along interlobular septa or within major and minor fissures. IPLs histologically resemble classic lymph nodes with well-developed cortical and medullary areas.12,14 Sinus histiocytes frequently contain abundant anthracosilicotic pigment and silicotic nodules with calcifications may form (Fig. 120-2). In patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), IPLs may be involved and, rarely, primary lung carcinoma or carcinoma from a nonpulmonary site can metastasize to IPLs. Most importantly, IPLs can be clinically, radiographically, and cytologically mistaken for malignancy.15,16 Although fine-needle aspirates can exclude the possibility of carcinoma, an erroneous diagnosis of lymphoma might be considered.
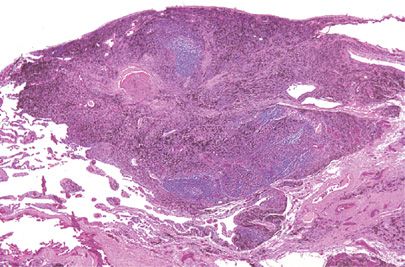
Figure 120-2 Intrapulmonary lymph node. Subpleural lymph nodes often accumulate carbon pigment and become fibrotic. The radiologic differential diagnosis includes carcinoma, while a fine-needle aspirate sample may mimic malignant lymphoma. Hematoxylin and eosin, 4× original magnification.
GENERAL CONSIDERATIONS
Although internists, pulmonologists, and radiologists often diagnose pulmonary diseases on the basis of clinical and radiographic findings and rely on tissue samples merely for confirmation, lymphoid lesions of the lung are often unsuspected diagnoses rendered by pathologists. Yet clinical and radiologic studies are essential in the interpretation of all lesions. Often, light microscopic considerations can be excluded purely on the basis of radiographic findings. For example, whereas pulmonary lymphomas can be localized or diffuse, lymphoid interstitial pneumonia (LIP) is always bilateral and diffuse.
Although fine-needle aspirate biopsies and transbronchial biopsies supplemented with ancillary studies may suffice for a diagnosis of malignant lymphoma, architectural and cytologic variability necessitates generous sampling. Since cellular monotony is not the sole criterion for malignancy, pulmonary lymphoid lesions often require wedge biopsies for diagnosis. Whereas sheets of uniform cells may be diagnostic of lymphoma, many malignant processes such as Hodgkin lymphoma (HL) and T-cell lymphoma are polymorphous and admixed with inflammatory cells. Secondary changes and biopsy-related artifacts may also confound the interpretation of a small sample.
Larger samples also allow for low-magnification pattern recognition. A “lymphatic distribution” may be seen in nonlymphoid processes such as sarcoidosis, yet is most striking in lymphoproliferative lesions reflecting the homing of lymphoid cells to endogenous pulmonary lymphatic routes. Although malignant lymphoid processes usually obliterate underlying lung architecture, diffuse alveolar septal expansion with lymphoid cells without a beaded lymphangitic pattern tends to represent an inflammatory rather than neoplastic process.
In addition to histologic examination, immunophenotyping is routinely performed and has become indispensable in diagnosing and classifying lymphoid lesions of the lung. Immunohistochemical studies can be reliably performed on formalin-fixed, paraffin-embedded tissue, whereas flow cytometry is useful for demonstrating immunoglobulin light-chain restriction. Aberrant antigen expression by either method also allows for subclassification. Polymerase chain reaction (PCR) may be required in up to 20% of cases to prove clonality.17 Either rearrangement of the immunoglobulin heavy-chain gene joining region (JH) or the T-cell receptor γ-chain gene (TCR-γ) can be investigated. Lastly, chromosomal abnormalities indicative of specific lymphomas such as t(14;18) translocation and bcl-2 gene rearrangement in follicular lymphoma or t(11;14) translocation and cyclin D1 (bcl-1) gene rearrangement in mantle cell lymphoma may be helpful.
Given the complex evaluation required of these lesions, it is incumbent upon the clinician to deliver the fresh tissue sample to the pathologist along with his or her concern for lymphoma. Although formalin-fixed or B5 fixed, paraffin-embedded samples may suffice for diagnosis and immunohistochemical evaluation, cell suspensions and fresh frozen tissue are required for flow cytometry, cytogenetics, and many molecular studies.
Lastly, although clonality indicates malignancy, clonality in pulmonary lymphoid lesions may not predict clinical outcome. Many pulmonary lymphomas have long indolent courses with 10-year survival rates of more than 80% yet LIP, a polyclonal process, is progressive with one-third of affected patients dying of end-stage pulmonary fibrosis.18,19
REACTIVE LYMPHOID PROCESSES
Although the terminology used to describe inflammatory processes of the lung has remained relatively static over the past 25 years, immunohistochemistry and molecular genetic analysis have redefined diagnostic criteria. Processes arising from BALT include nodular lymphoid hyperplasia (NLH), follicular bronchitis/bronchiolitis (FBB), diffuse lymphoid hyperplasia (DLH), and LIP. Pulmonary hyalinizing granuloma (PHG) is included in this section for historical reasons only. Although the lesions are best considered either localized or diffuse, clinicopathologic entities including Castleman disease (CD) can manifest with either distribution.
 LOCALIZED REACTIVE LYMPHOID PROCESSES
LOCALIZED REACTIVE LYMPHOID PROCESSES
Localized reactive lymphoid processes, including nodular lymphoid hyperplasia, pulmonary hyalinizing granuloma, amyloidosis, and light chain deposition disease are discussed below.
Nodular Lymphoid Hyperplasia
Known to previous generations of pulmonologists and hematopathologists as “pseudolymphoma,” this reactive process was recognized as a form of BALT hyperplasia in the mid-1980s.20 Although NLH was once considered to be quite common, ancillary studies have convincingly demonstrated that most cases were actually low-grade lymphomas.21 Thus true NLH is now considered a legitimate but exceedingly rare polymorphous nodular lymphoid lesion of the lung.22 Neutrophilic microabscesses and foreign body giant cells found in several reported cases indicate that the lesion may be the result of an inflammatory stimulus. Interestingly, immunohistochemical studies have documented B-cell lymphomas of BALT within pre-existing reactive masses of BALT, such that one could, in the most general sense, consider NLH a precursor lesion.11,20
Most affected individuals are middle aged with a nearly equal gender incidence.22,23 Patients are usually asymptomatic, although a small percentage may have autoimmune diseases such as systemic lupus erythematosus or Sjögren syndrome, or polyclonal hypergammaglobulinemia. Lesions usually appear as solitary subpleural radiographic nodules with air bronchograms, but several nodules or localized infiltrates can be seen, the latter finding serving as a reminder that the distinction between nodular and diffuse processes is arbitrary.24 Up to one-third of cases feature regional lymphadenopathy.
Excised tan-white rubbery to firm nodules measure from 0.6 to 6.0 cm. Histologically, the well-demarcated lesions may feature slight extension along alveolar septa and central scarring. Normal lung parenchyma is overrun by large reactive germinal centers with well-preserved mantle zones, and lymphoepithelial lesions are not seen (Fig. 120-3). Interfollicular areas are filled with plasma cells and mature lymphocytes. The follicles are clearly reactive with a variety of cell types, mitoses, and macrophages. Regional lymph nodes often feature reactive follicular hyperplasia.
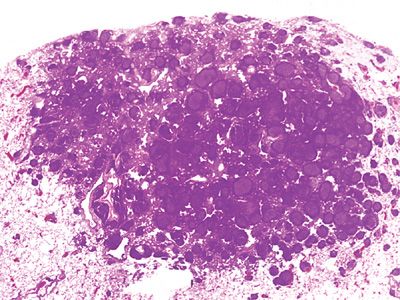
Figure 120-3 Nodular lymphoid hyperplasia. The lesion is composed of benign reactive germinal centers. Although radiographically nodular, germinal centers spill out into surrounding lung. Immunohistochemistry is required to exclude a diagnosis of malignant lymphoma. Hematoxylin and eosin, 1× original magnification.
Immunohistochemical studies demonstrate a mixture of B- and T-cells. The B-cells express both κ and λ light chains, that is, lack light chain restriction. Aberrant B-cell staining for CD5, CD23, or CD43 is not seen, and the germinal centers do not stain with bcl-2.22,25 Immunoglobulin heavy chain gene rearrangement or evidence of t(14;18) breakpoints is not observed. These ancillary findings are of the utmost importance since light microscopy alone may not differentiate NLH from an extranodal marginal zone B-cell lymphoma of MALT. The latter usually features infiltrative growth, but may have reactive follicles and polytypic plasma cells with only a focal monotypic cell population.
Surgical excision is usually curative, although a small percentage of patients develop local recurrences at the original surgical site. Neither systemic spread nor death has been reported.22,25
Pulmonary Hyalinizing Granuloma
PHG is neither a lymphoid nor granulomatous lesion per se, but an ever-present lymphoid component allows for its discussion in a hematolymphoid chapter. This peculiar fibrosing process shares clinicopathologic and morphologic features with sclerosing mediastinitis, inflammatory pseudotumor of the orbit, Riedel thyroiditis, and idiopathic retroperitoneal fibrosis.26 In fact, approximately one-fourth of cases feature concomitant mediastinal or retroperitoneal disease.27,28 The etiology is unknown but it has been speculated to represent either an autoimmune phenomenon or exaggerated host response to mycobacteria or fungi.
Age at presentation ranges from 24 to 77 years and women are affected twice as often as men. Most patients present with mild symptoms including cough, shortness of breath, fever, and fatigue but up to 25% of reported individuals are asymptomatic. Laboratory studies include positive antinuclear antibodies, rheumatoid factor, antineutrophil cytoplasmic antibodies, and Coombs-positive hemolytic anemia.28 Elevated serum or tumor immunoglobulin G4 (IgG4) levels are rarely reported.29,30 Skin testing often demonstrates exposure to Mycobacterium tuberculosis or Histoplasma capsulatum, but cultures and stains for microbes are negative. Radiographs reveal less than 4.0 cm bilateral and multilobar ill-defined homogeneous nodules that resemble metastases.31 Unilateral and solitary cases measuring up to 15 cm have been reported. Although not common, focal central irregular calcification may suggest metastatic bone-forming neoplasms. Central cavitation is rare.
Lesions are sharply circumscribed white-tan rubbery masses composed of irregular concentric whorls of hyalinized collagen encasing vessels and airways (Fig. 120-4A).27,28 The center of the lesion is paucicellular, whereas peripheral thick collagen bands are separated by mature T-lymphocytes, plasma cells, fibroblasts, and occasional giant cells (Fig. 120-4B). Blood vessels may feature transmural inflammation without necrosis. Microscopic calcifications can be seen, but granulomas are not present despite the designation PHG. The tumoral interface with lung parenchyma features reactive germinal centers, whereas adjacent lung may feature organizing pneumonia and hyperplasia of BALT. The morphologic differential diagnosis includes rheumatoid nodule, amyloidosis, granulomatosis with polyangiitis (Wegener granulomatosis), malignant lymphoma, inflammatory myofibroblastic tumor, and infections.29 Clinicians should not be comfortable with a diagnosis of PHG rendered on anything less than a completely removed lesion.
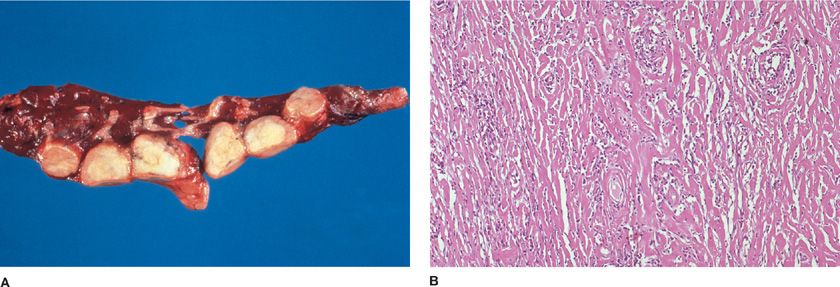
Figure 120-4 Pulmonary hyalinizing granulomas. A. Multiple well-circumscribed tan firm nodules can compromise respiratory function. B. Pink hyalinized collagen bands encircle vessels. Scant benign lymphoid infiltrates percolate between the collagen. Hematoxylin and eosin, 10× original magnification.
PHG tends to enlarge slowly and does not recur after surgical resection. However, growth of unresected or unresectable nodules can lead to respiratory compromise.28
Amyloidosis and Light Chain Deposition Disease
Immunoglobulin light chains can accumulate in many tissues in different forms, depending on the underlying condition and particular organ cytoskeleton. Clinically recognizable disease in the lung can manifest as tracheobronchial disease, solitary or multiple nodules, cystic lung disease, or in a diffuse interstitial parenchymal pattern.32,33 Most cases of diffuse light chain deposition in the lung are part of multiorgan involvement, are associated with plasma cell dyscrasia, have dismal clinical outcomes, and are not discussed further in this section.
Solitary and multiple amyloidoma are seen most often in older individuals, with a mean age of 67 years.32 Central and solitary lesions are often incidental findings but airway or visceral pleural distortion may produce cough, hemoptysis, or pleuritic chest pain. A radiographic diagnosis can be suggested when calcification or ossification is noted (20%–50% of the time); otherwise, the clinical impression is that of a neoplasm.34
Serum or urine monoclonal proteins are found in 10% of patients, and this lung pathology may be associated with lymphoproliferative diseases such as benign monoclonal gammopathy of undetermined significance, Sjögren syndrome, Crohn disease, NLH, LIP, extranodal marginal zone B-cell lymphoma of MALT, or multiple myeloma.32,35–37 Solitary amyloidoma without underlying blood dyscrasias probably represent a hyperimmune response to unknown antigens.
Waxy hard gritty and yellow-tan nodules measure up to 15 cm and are composed of amorphous eosinophilic hyaline material that obliterates lung parenchyma but spares many arterioles (Fig. 120-5). Lymphocytes, plasma cells, and multinucleated giant cells percolate through the amyloid, but the infiltrate is most dense at the periphery of the nodules. Calcification and ossification with secondary marrow space formation is common. Congo red staining examined by polarizing microscopy reveals lesional apple-green birefringence. Immunohistochemical studies usually demonstrate λ light-chain composition and negative immunoreactivity for amyloid A and transthyretin.37,38 Plasma cells are most often polytypic; however, small foci of monoclonal plasma cells within foci of polytypic plasma cells have been identified.39 Ultrastructurally, amyloid is composed of disorderly nonbranching hollow-core 8- to 10-nm fibrils. These extracellular deposits of chemically diverse proteins form a three-dimensional twisted β-pleated sheet. Resected lesions may not demonstrate malignancy, but patients should be screened for underlying monoclonal B-cell proliferations including multiple myeloma and overt B-cell malignancies.
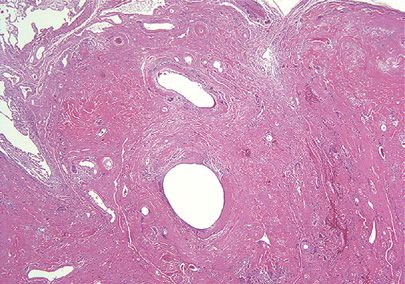
Figure 120-5 Nodular amyloidosis. Amorphous pink material replaces air spaces and overruns airways. In the absence of Congo red apple-green birefringence one should consider the possibility of nodular light chain deposition. Hematoxylin and eosin, 4× original magnification.
In stark contrast to nodular amyloidosis, pulmonary light chain deposition disease (LCDD) is associated with an underlying blood dyscrasia or renal failure in more than 50% of affected individuals.40–42 Most patients have free κ monoclonal light chains (IgG, IgA, and IgM in decreasing order) in their urine or serum.43 The clinical and light microscopic appearance of LCDD is similar to amyloid, and one might mistake a case lacking the characteristic Congo red staining as simply a poorly stained example of amyloid. These light chain deposits are composed of amorphous granular or globular electron dense material.43 One should consider this entity when dealing with a nonamyloidotic deposit given its strong association with lymphoid malignancies including but not limited to multiple myeloma and renal failure.43,44
 DIFFUSE REACTIVE LYMPHOID PROCESSES
DIFFUSE REACTIVE LYMPHOID PROCESSES
Although NLH most likely represents a local response to an extrinsic stimulus and is clinically relevant owing to its radiographic appearance as a coin lesion and morphologic similarity with extranodal marginal zone B-cell lymphoma of MALT, the DLHs, FBB, and LIP represent a continuum of BALT hyperplasia often seen in patients with systemic diseases (Table 120-1).20,25,45 The extent of lung involvement is due in large part to host immune factors.46 Although thoracoscopic or open lung biopsies are required to establish these diagnoses and exclude other processes, including interstitial lung diseases and malignant lymphoma, morphology does not suggest etiology.
Follicular Bronchitis/Bronchiolitis
DLH restricted to the walls of airways and peribronchial tissue is referred to as FBB. FBB is often seen in individuals with bronchiectasis, chronic infections, and chronic obstructive pulmonary diseases, including asthma.45,47 The entity may also be seen in association with connective tissue diseases, congenital or acquired immunodeficiencies, or bone marrow transplantation; or as a manifestation of a hypersensitivity reaction.48,49 When the process spreads along lymphatic routes of the pulmonary lobule some prefer the designation DLH instead of FBB.
Patients with FBB in association with connective tissue disease are usually in their forties and most often suffer from rheumatoid arthritis (RA) or Sjögren syndrome.50,51 Patients with immunodeficiency syndromes such as acquired immunodeficiency syndrome (AIDS), common variable immunodeficiency, IgA deficiency, and Evans syndrome present in childhood, whereas those with hypersensitivity syndromes are usually in their sixth decade of life. Patients may also suffer with chronic low-grade infections such as mycoplasma, chlamydia, or Epstein–Barr virus (EBV).45
Individuals with FBB present with dyspnea, cough, and fever; some may have recurrent pneumonia or weight loss. Pulmonary function tests reveal restrictive, obstructive, or normal patterns.47 Those with RA often have a very high rheumatoid factor on the order of 1:640 to 1:2560. Peripheral eosinophilia may be noted in those with hypersensitivity syndromes. Arterial blood gases show arterial hypoxia with a widened AaPO2 gradient and hypocapnia. Chest radiographs feature bilateral diffuse reticular and nodular opacities, whereas HRCT show up to 12-mm centrilobular and peribronchial nodules with or without areas of ground-glass opacity.24,52
Gross pathology demonstrates numerous minute (1- to 2-mm) nodules adjacent to airways. Microscopically, nodular aggregates of B-cell rich lymphocytes and plasma cells with reactive germinal centers expand bronchial and/or bronchiolar submucosa and budge into and permeate overlying epithelium (Fig. 120-6). Rare T-cells wander beyond the follicles into adjacent alveolar septa.25 Smaller airway lumens are distorted and narrowed predisposing to mucostasis and subsequent infections. Lymphoid follicles along interlobular septa and beneath the pleura represent a more diffuse form of lymphoid hyperplasia and warrant the descriptive diagnosis DLH.
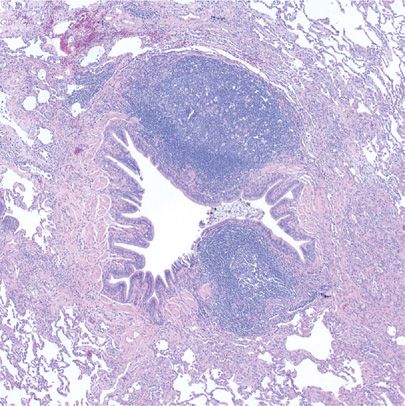
Figure 120-6 Follicular bronchitis/bronchiolitis. Reactive germinal centers expand airway submucosa and compress the lumen. Mucus accumulation often leads to infection and bronchiolectasis. Alveolar parenchyma is spared. Hematoxylin and eosin, 4× original magnification.
Treatment of FBB with corticosteroids has variable results.47,48 Those with peripheral eosinophilia are reportedly steroid responsive.
Lymphocytic Interstitial Pneumonia
Although LIP is included in the American Thoracic Society/European Respiratory Society (ATS/ERS) classification of idiopathic interstitial pneumonias, this multifactorial but rarely idiopathic process represents the most florid form of BALT hyperplasia and may be difficult to differentiate from florid FBB and low-grade malignant lymphoma.53 Almost all patients with LIP have immunologic disorders, dysproteinemias, or viral infections, including EBV and, especially in children, human immunodeficiency virus (HIV).54–57 The age and sex distribution reflect these different populations. This clinicopathologic process is rare in the HIV-negative population, but represents a pulmonary manifestation of chronic graft-versus-host (GVH) disease in bone marrow transplant patients.54 A strong association with Sjögren syndrome is also noted.58 Most HIV-negative patients are middle-aged Caucasian women.
The clinical presentation includes cough and/or dyspnea in addition to symptoms and signs related to underlying diseases. More than 60% of patients have dysproteinemias, which can precede the onset of LIP or occur any time during the clinical course.54,59,60 Most of these cases are associated with hypergammaglobulinemia; only 10% of cases are associated with hypogammaglobulinemia. A monoclonal spike on serum immunoelectrophoresis suggests a diagnosis of lymphoma rather than LIP. Pulmonary function tests reveal reduced lung volumes and diffusing capacity for carbon dioxide (DLCO), and hypoxia is common.54 Bronchoalveolar lavage (BAL) analysis shows an increased percentage of lymphocytes. Chest radiographs demonstrate bilateral reticular and nodular opacities, ground-glass opacities, and parenchymal consolidation with lower lung zone predilection. Computed tomography demonstrates diffuse ground-glass opacities, ill-defined centrilobular nodules, bronchovascular and interlobular thickening, and scattered less than 3.0-cm thin-walled cysts.61–63
The lungs are typically firm and tan-gray and end-stage cases feature honeycomb change with subpleural cysts. Although the radiographs and gross appearance suggest parenchymal consolidation, histologically LIP shows a diffuse prominently interstitial infiltrate of small lymphocytes, plasma cells, larger mononuclear cells, and histiocytes (Fig. 120-7A). Although these infiltrates are centered on airways, vessels, and interlobular septa, and include peribronchiolar lymphoid follicles, infiltration into alveolar septa is always present and distinguishes LIP from FBB (Fig. 120-7B). Small nonnecrotizing granulomas, reactive germinal centers, and infiltration into overlying respiratory epithelium are often seen, whereas lymphocytes frequently spill into alveolar spaces. In long-standing lesions, hyaline, collagen, or even amyloid widens the interstitium leading to honeycomb fibrosis. The lymphoid follicles largely consist of cytologically bland B-cells, whereas the interstitial lymphocytes are mostly T-cells. This pattern suggests that the lung can function like a giant lymph node.25 Immunoglobulin heavy chain restriction or gene rearrangement is lacking. In addition to malignant lymphoma, nonspecific interstitial pneumonia (NSIP) and infections including Pneumocystis should be excluded.
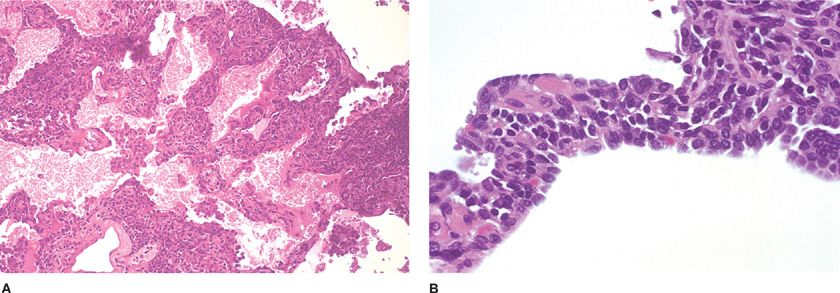
Figure 120-7 Lymphocytic interstitial pneumonia. A. Diffuse alveolar septal expansion with lymphocytes is usually a manifestation of underlying disease. Hematoxylin and eosin, 50× original magnification. B. Benign lymphocytes and plasma cells interfere with gas exchange. The morphologic differential diagnosis includes pneumocystis infection. Hematoxylin and eosin, 60× original magnification.
Given the rarity of LIP, controlled treatment trials have not been undertaken. Corticosteroids are the primary therapy in addition to other immunosuppressive agents, such as cyclophosphamide and chlorambucil, with variable results. One-third of patients have resolution, one-third stabilize, and the remaining third progress.18,54,60 Nonresponders often die of therapy-related infections, but occasional individuals die of end-stage pulmonary fibrosis. Lymphomatous transformation is very unusual; older reports of such most likely represented malignant lymphomas from the start.64
In patients with HIV infections or AIDS, LIP is part of a spectrum of lymphoid proliferations with virtually identical morphologies, but differing clinical presentations. Individuals with so-called diffuse infiltrative lymphocytosis syndrome (DILS) featuring sicca syndrome with increased numbers of circulating CD8+ T-cells in the blood, generalized lymphadenopathy, and enlarged parotid glands, are at high risk of developing LIP.65
LIP is most common is HIV-positive children and is a CDC category B indicator condition in children younger than age 13.66,67 In fact, up to 17% of HIV-positive children have LIP.68 Most present in their second or third year with lung infiltrates, failure to thrive, and increasing respiratory distress. The chest radiograph shows a diffuse micronodular or linear interstitial pattern with hilar and mediastinal widening.69 Therapy is uncertain, response to steroids is unpredictable, and mean survival is 33 months.70
In HIV-positive adults, LIP is quite rare, and tissue sampling is required for diagnosis.57 Most patients present with generalized lymphadenopathy and polyclonal hypergammaglobulinemia. BAL samples feature lymphocytes with CD8+ cells comprising up to 90% of the lymphoid cells.71 Histologically, the lymphoid infiltrates are predominantly T-cells with few plasma cells. Germinal center formation is not a frequent finding. HIV-positive adults with LIP rarely die of the process, but rather of other AIDS-related diseases.
Castleman Disease
CD, the eponymous term for angiofollicular lymph node hyperplasia, encompasses two clinically and pathologically distinct entities. Solitary lesions usually feature hyaline-vascular (HV-CD) morphology, whereas multicentric disease almost always has a plasma cell pattern (PC-CD).72
Solitary CD is an uncommon form of lymphoid hyperplasia that usually presents as an incidental mediastinal mass in asymptomatic young to middle-aged individuals of either gender.73 Pleural, chest wall, and extrathoracic involvement have been reported, but pulmonary parenchymal disease is a true rarity and most reported cases likely represent nodal rather than true pulmonary disease (Fig. 120-8A).73–78 Solitary HV-CD lesions are usually asymptomatic or rarely cause compression-related symptoms.
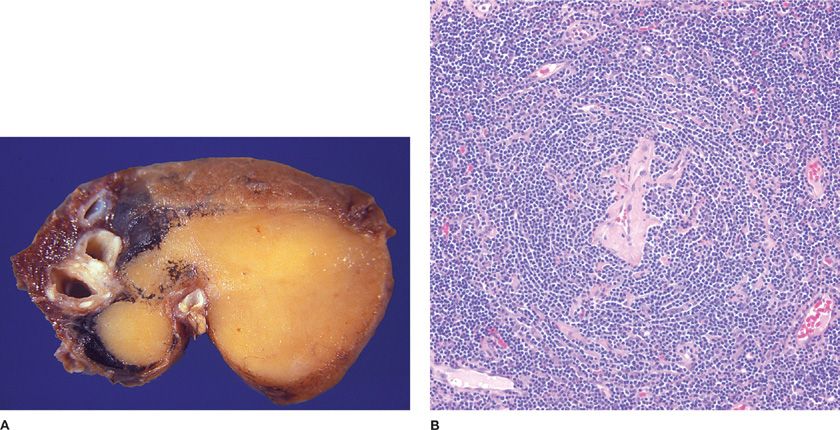
Figure 120-8 Solitary Castleman disease. A. Involved peribronchial lymph nodes are often mistaken for pulmonary parenchymal disease. Airway and vessel distortion may cause symptoms. B. Solitary lymph nodes and rare lung lesions feature hyalinevascular morphology. Small germinal centers are penetrated by hyalinized venules. The follicle mantle zone rings the burnt-out center in an onionskin pattern. Hematoxylin and eosin, 20× original magnification.
Ninety percent of solitary CDs feature hyaline-vascular morphology, whereas the remainder are the plasma cell variant. HV-CD lymph nodes are enlarged and feature prominent lymphoid follicles with small atrophic germinal centers penetrated by hyalinized venules with plump endothelial cells originating in the interfollicular zone (Fig. 120-8B). Expanded mantle zones have concentric rings of lymphocytes imparting an onionskin appearance. TdT+ T-lymphoblastic populations are increased in this entity.79 The solitary plasma cell variant only involves lymph nodes and has not been reported in the lung. Lymph nodes are hyperplastic with enlarged germinal centers and sheets of interfollicular plasma cells. Clinical suspicion of malignancy typically prompts surgical resection of solitary lesions. Excision results in the disappearance of any symptoms.80 Neoadjuvant anti-CD20 monoclonal antibodies can shrink lesions prior to surgery.81
Multicentric Castleman disease (MCCD) is best considered a virus-driven polyclonal lymphoproliferative process that shares virtually no features with solitary CD.72,82,83 In general, MCCD presents in the fourth and fifth decades of life but earlier in HIV-positive individuals.82,84,85 An increased incidence is noted in the highly active antiretroviral therapy (HAART) era.86 Signs and symptoms include fever, sweating, malaise, anemia, lymphadenopathy, hepatosplenomegaly, ascites, and pleural and pericardial effusions. MCCD appears to be a consequence of infection with human herpesvirus 8 (HHV-8), which in turn promotes production of interleukin-6 (IL-6).72 Laboratory abnormalities include an elevated erythrocyte sedimentation rate, polyclonal hypergammaglobulinemia, and bone marrow plasmacytosis that may lead to pancytopenia.87 A chronic demyelinating polyneuropathy may present as part of POEMS syndrome (Crow–Fukase disease).88 Patients may also develop non-HL. HIV-positive patients with MCCD have a greater likelihood of pulmonary involvement.89
Pulmonary histomorphology correlates with HRCT scan findings of peribronchovascular interstitial thickening and centrilobular nodules (Fig. 120-9A).90,91 Polyclonal peribronchiolar lymphoplasmacytic infiltrates with focal extension into interlobular and alveolar septa may rarely be associated with honeycomb change (Fig. 120-9B). HHV-8 can almost always be demonstrated in lung samples, including BAL, by PCR and in situ hybridization with an HHV-8 probe.92 Although MCCD shares many features with LIP, the plasma cell–rich nature of the infiltrate and presence of HHV-8 discriminate between the two.
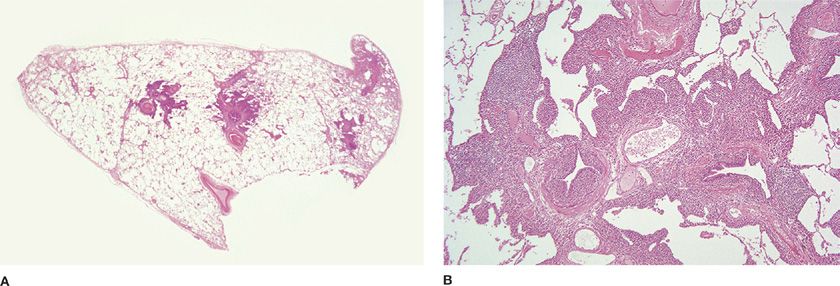
Figure 120-9 Multicentric Castleman disease. A. The bronchocentric nature of this KSHV/HHV8-driven systemic process is apparent at scanning microscopy. Hematoxylin and eosin, 1× original magnification. B. The lymphoplasmacytic infiltrate is confined to the pulmonary interstitium. Honeycomb change may develop. Hematoxylin and eosin, 10× original magnification. (Glass slides used with permission of Dr. J. English, University of British Columbia and Vancouver Hospital, Vancouver, BC.)
Treatment is primarily nonsurgical but survival beyond 5 years is rare.87 Although splenectomy may provide brief relief, chemotherapeutic regimens with or without immunotherapy, including anti–IL-6 and anti-CD20 monoclonal antibodies, appear to induce the longest remissions.74,84,93 An 85% 1-year survival rate is now reported.93
Immunoglobulin G4–related Lung Disease
IgG4-related disease is a recently recognized fibroinflammatory condition with diverse organ manifestations linked by a unique histologic appearance.94 As such, IgG4-related disease mirrors sarcoidosis. Despite a highly variable clinical presentation, the common histologic appearance, elevated serum, and tissue IgG4 levels along with a clinical response to immunosuppression warrant the creation of the broad yet poorly understood entity.95 While most of the initial observations were made in individuals with autoimmune pancreatitis and/or lacrimal and salivary gland disease, the process is now recognized in almost every organ system.96–98 General and organ-specific diagnostic criteria have been developed.95,99 IgG4-related pulmonary disease likely accounts for many fibroinflammatory conditions of unknown etiology including but not limited to inflammatory pseudotumor, interstitial pneumonia, and fibrosing (sclerosing) mediastinitis.26,100 Some patients with autoimmune idiopathic pancreatitis also have lung manifestations.101
Most patients with lung disease are males in their seventh decade of life.102 One half of patients have respiratory symptoms such as cough, exertional dyspnea, hemoptysis, and/or chest pain. Constitutional symptoms are uncommon.103 No risk factors are recognized. Radiographic appearances vary and correlate with histomorphologic findings.104 Solid nodules, bronchovascular, alveolar interstitial, pleural, and airway patterns may be seen, alone or in various combinations.102 The three major histopathologic features are (1) dense lymphoplasmacytic infiltrate, (2) fibrosis, arranged at least focally in a storiform pattern, and (3) obliterative phlebitis (Fig. 120-10).95 IgG4-related pulmonary disease may lack distinct storiform fibrosis while pulmonary arteries, in addition to veins, may be involved.105 Furthermore, vascular changes without vessel lumen obliteration, concentric bronchiolar inflammatory infiltration with germinal centers, and/or prominent eosinophils may be seen.106 Not surprisingly, histomorphologic findings can be considered highly suggestive, probable, or insufficient.95 Unfortunately, the major findings are also seen in a myriad of entities, including but not limited to infections and organizing injuries related to, for example, granulomatosis with polyangiitis (Wegener granulomatosis).107
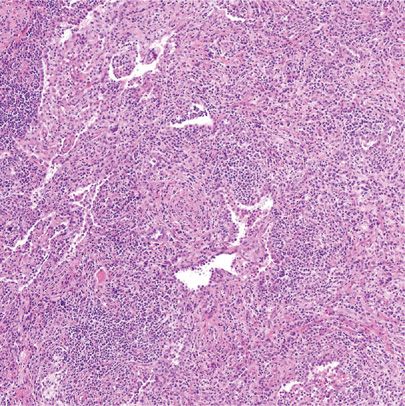
Figure 120-10 Immunoglobulin G4–related lung disease. Pulmonary parenchyma is practically overrun with lymphoplasmacytic infiltrates and developing fibrosis. Residual airspaces contain histiocytes. Hematoxylin and eosin, 10× original magnification. (Used with permission of Dr. W. Travis, Memorial Sloan Kettering Cancer Center, New York, NY.)
Tissue and serum IgG4 and IgG levels serve as supplemental data since a diagnosis is based on light microscopic findings. Immunohistochemical stain evaluation requires that one count the three 40x microscopic fields with the highest number of IgG4+ plasma cells and also count the number of IgG+ plasma cells in those same areas. According to the consensus statement, >50 IgG4+ cells per high-power field and >20 IgG4+ cells per high-power field are adequate cutoffs in surgical and nonsurgical lung biopsies, respectively.95 The IgG4+/IgG+ plasma cell ratio should be >40%. However, other lung processes including idiopathic NSIP, usual interstitial pneumonia, connective tissue disease-associated interstitial pneumonias as well as inflammatory myofibroblastic tumor may have increased IgG4+ cell counts.108 Elevated serum IgG4 levels are noted in most patients, although other respiratory, hepatobiliary tract, and connective tissue diseases may be associated with high serum levels.103,109
Standard therapy is not well delineated but most patients respond to glucocorticoids within weeks to months.110,111 Resistant cases may improve with anti-CD20 monoclonal antibody therapy.112 Relapses are common after therapy cessation. An increased risk for non-HL is postulated.113
MALIGNANT LYMPHOID LESIONS
Within the hematopathology field, past decades will probably be best remembered for the myriad of classification schemes and ever-changing nomenclature. Thankfully, the current WHO classification represents a consensus list of lymphoid neoplasms that appear to be distinct clinical entities.1 Although complex, this scheme is reproducible among trained pathologists. Diagnoses are based on clinical, morphologic, immunophenotypic, and genetic features and not simply on morphologic, immunophenotypic, or even clinical subtleties. B-cell neoplasms, T- and NK-cell neoplasms, and HL are subgrouped according to lineage and stage of differentiation. Within this general context one can understand the practicality of a seemingly cumbersome diagnosis, such as pulmonary extranodal marginal zone B-cell lymphoma of MALT type, given the belief that these lymphomas arise from acquired BALT.
 PRIMARY PULMONARY NON-HODGKIN LYMPHOMA
PRIMARY PULMONARY NON-HODGKIN LYMPHOMA
Although more than half of patients with nodal lymphoma have lung involvement, primary pulmonary lymphomas comprise less than 0.5% of primary lung neoplasms.114,115 Furthermore, the most common primary lung lymphoma, marginal zone non-HL of MALT origin, represents less than 10% of extranodal lymphomas.116 Non-HLs are considered lung primaries when the lung is the major site of disease at the time of diagnosis.117,118 Thus, up to 20% of these lung lymphomas involve hilar and/or mediastinal lymph nodes. Pulmonary lymphomas have a range of morphologies and clinical aggressiveness such that separating the tumors into low- and high-grade categories is a dangerous oversimplification. Although most lung non-HLs differ from nodal lymphomas and are thought to have their origin in BALT, traditional nodal non-HLs, such as follicular lymphoma, mantle cell lymphoma, diffuse large B-cell lymphoma, peripheral T-cell lymphoma, and CD30+ anaplastic large cell lymphoma, also present as pulmonary primaries (Table 120-2).19,119–122
T-cell and NK-cell neoplasms
Mature T-cell and NK-cell neoplasms
Mycosis fungoides
Sézary syndrome
Peripheral T-cell lymphoma, unspecified
Angioimmunoblastic T-cell lymphoma
Anaplastic large cell lymphoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


