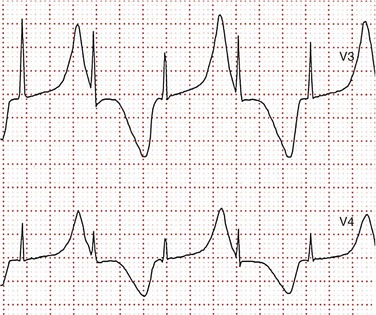
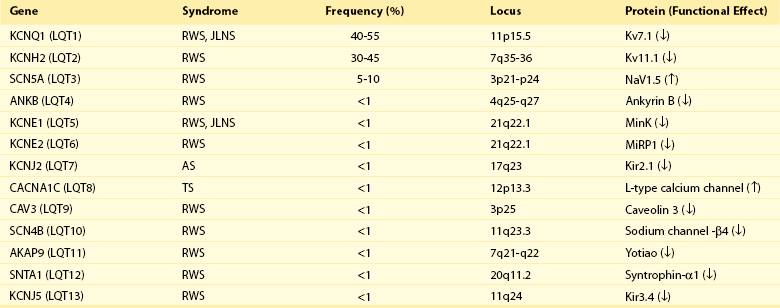
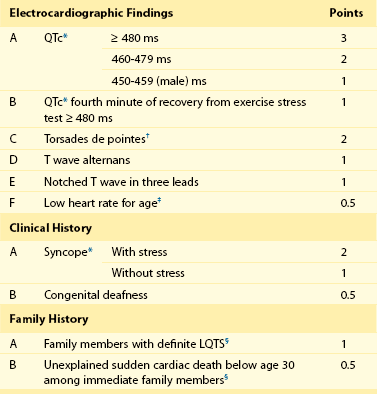
93 Congenital long QT syndrome (LQTS) is a relatively uncommon but important clinical disorder. Since 1975,1 under the unifying name of long QT syndrome, it includes two hereditary variants: Jervell and Lange-Nielsen syndrome (J-LN) associated with deafness and the Romano-Ward syndrome (R-W), which is not associated with deafness. Following the identification in 1995 to 1996 of the first three LQTS genes2—associated with the most frequent variants LQT1, LQT2, and LQT3—several others were identified. Unfortunately, haste or enthusiasm has created a terminology problem because some of these genes cannot be truly regarded as responsible for LQTS. What have been called LQT4 and LQT7 (Andersen-Tawil syndrome) are complex clinical disorders in which a modest prolongation of the QT interval is only a secondary epiphenomenon and should not be regarded as part of LQTS. LQT5, LQT6, and LQT8, although rare, are part of LQTS. For the remaining 5 variants (LQT9 to LQT13) there are only preliminary descriptions.2 As shown in Table 93-1, the main characteristic of all the LQTS genes identified thus far is to encode cardiac ion channel subunits or proteins, thus modulating ionic currents. Table 93-1 From Schwartz PJ, Crotti L, Insolia R: Long QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol 5:868–877, 2012. The SCN5A gene encodes the protein of the cardiac sodium channel. In vitro expression studies3 showed that LQTS-SCN5A mutations produce the LQTS phenotype by inducing a gain of function leading to an increase in the Na+ inward current, which prolongs action potential duration. The prevalence of LQT3 among genotype-positive LQTS patients is 10% to 15%. LQT8 is a rare variant characterized by marked QT interval prolongation, often presenting with a 2 : 1 functional atrioventricular block, macroscopic T wave alternans, and syndactyly. LQT8 is highly malignant, and 10 of the 17 (59%) children reported by Splawski et al.4 died at a mean age of 2.5 years. Some children with Timothy syndrome also had congenital heart diseases, immune deficiency, intermittent hypoglycemia, cognitive abnormalities, and autism. Molecular screening identified a missense mutation (G406R) in the voltage-gated calcium channel gene (CACNA1c) in all probands analyzed. G406R produces sustained inward Ca++ currents by causing nearly complete loss of voltage-dependent inactivation4. In the heart, prolonged Ca++ current delays cardiomyocyte repolarization and increases risk of arrhythmia. Although it was suggested in 1975 that LQTS “could be more unrecognized than rare,”1 only recently a data-driven indication of the prevalence of LQTS became available and came from the largest prospective study of neonatal electrocardiography ever performed5, which involved 44,596 infants 3 to 4 weeks old. Among them, 1.4% had a QTc between 440 and 469 ms and 0.7 per 1000 had a QTc ≥ 470 ms, regarded as markedly prolonged by the European Task Force on Neonatal Electrocardiography.5 In the latter group (n = 31), more than 90% of infants underwent molecular screening, and LQTS disease-causing mutations were found in 13 of 28 infants (46%).5 Because almost 50% of the infants with QTc ≥ 470 ms (0.7/1000) are affected by LQTS, and because at least some of the infants with QTc between 440 and 469 ms are also likely to be affected, it follows that the prevalence of LQTS must be close to 1 per 2500 at least. This does not include the silent mutation carriers (QTc < 440 ms), a group ranging between 10% and 36% according to genotype.6 This is the first and only time that the prevalence of a cardiac disease of genetic origin has been quantified based on actual data. The syncopal episodes are the result of torsade de pointes (TdP) often degenerating into ventricular fibrillation. Although most patients develop their symptoms under stress, sometimes these life-threatening cardiac events occur at rest. The reasons for these different patterns remained obscure until molecular biologists were able to distinguish among different genotypes. In 1995, a tiny group of genotyped individuals LQT2 patients appeared to be at higher risk during emotional stress,7 whereas LQT3 patients suffered their events mostly at rest or during sleep; this fostered a large and targeted study8 that shed the light on this issue of major clinical relevance. In 670 patients with LQTS of known genotype and who had suffered symptoms (e.g., syncope, CA, sudden death), Schwartz et al.8 examined possible relations between genotype and the conditions (“triggers”) associated with the events.8 As predicted by their impairment on the IKs current (essential for QT shortening during increases in heart rate), most of the events in patients with LQT1 occurred during exercise or stress. Conversely, most of the events (including the lethal ones) in LQT2 patients occurred during emotional stress such as auditory stimuli (e.g., sudden noises, telephone ringing, especially while at rest), and most of the events of LQT3 patients occurred while they were asleep or at rest (Figure 93-1). Figure 93-1 Lethal cardiac events according to triggers and genotype. Numbers in parentheses are triggers, not patients. (Modified from Schwartz PJ, Priori SG, Locati EH, et al: Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 92:3381–3386, 1995.) Even in the postpartum period, genotype is important because risk is higher for LQT2 than for LQT1.2 The higher risk for women with LQT2 is partly related to sleep disruption, but hormonal changes could be involved as well. Indeed, a study performed during the menopause transition and postmenopausal periods (5 years before and after the age of onset of menopause, respectively) showed that women with LQT2 had an increased risk of cardiac events in both conditions (hazard ratio 3.38 and 8.10, respectively), whereas for women with LQT1 the onset of menopause was associated with a reduction in the risk for cardiac events (hazard ratio 0.19).9 The initial and understandable concept that QT prolongation was the essential cornerstone of LQTS was challenged when Schwartz and Crotti10 proposed that some patients might be affected by LQTS and nonetheless have a normal QT interval on the surface ECG. The validity of this unorthodox concept was proved by the existence of mutation carriers with a normal QT interval as a consequence of low penetrance.2,6 This concept has important practical and medicolegal implications because, for example, it no longer allows a cardiologist to state that a sibling of an affected patient with a normal QTc “is definitely not affected by LQTS.” Notched T waves are more frequent in symptomatic patients (81% versus 19%; P < .005).2 They probably reflect the presence of subthreshold early afterdepolarizations (EADs). Their appearance after exercise is markedly more frequent (85% versus 3%; P < 0.0001) among patients with LQTS than among healthy controls. It is worth recalling that notched T waves are not necessarily abnormal in children. In 1975, Schwartz and Malliani proposed that T wave alternans represents a characteristic ECG feature of LQTS, and this observation has been fully confirmed.10 Beat-to-beat alternation of the T wave, in polarity or amplitude, may be present at rest for brief moments, but it usually appears during emotional or physical stresses and can precede TdP (Figure 93-2); it is a marker of major electrical instability and identifies patients at particularly high risk. Its transient nature limits the possibility of observation; this is a rather gross phenomenon that should not go unnoticed when present. The observation of T wave alternans in a patient with LQTS receiving therapy strongly suggests the presence of a high degree of cardiac electrical instability, and it should prompt reassessment of therapy. In a large South African LQT1 founder population in which all the affected members carry the KCNQ1-A341V mutation, Brink et al.11 and Schwartz et al.12 provided the novel evidence that faster basal heart rates and brisk autonomic responses are associated with a greater probability of being symptomatic. Whereas among patients with a major arrhythmogenic substrate (QTc > 500 ms) basal heart rate is rather unimportant, among patients with a QTc ≤ 500 ms those in the lower tertile of heart rate were more frequently asymptomatic. Furthermore, relatively low values of baroreflex sensitivity—an index of the ability to respond with brisk increases in either vagal or sympathetic activity—were associated with a reduced probability of being symptomatic.12 This finding likely depends on the fact that patients with LQT1 have an impaired ability to shorten their QT interval during heart rate increases because of the mutation-dependent impairment in IKs, the current essential for QT adaptation. The lack of QT shortening during sudden heart rate increases favors the R-on-T phenomenon and initiation of ventricular tachycardia or ventricular fibrillation, whereas sudden pauses elicit early afterdepolarizations in patients with LQTS, which can trigger TdP. Blunted autonomic responses, revealed by relatively low values of baroreflex sensitivity (BRS), imply a reduced ability to change heart rate suddenly, which appears to be a protective mechanism for patients with LQT1. BRS is not frequently used in clinical practice; therefore, we recently assessed the value of another and simpler marker of reflex vagal activation (i.e., the heart rate reduction during the first minute of recovery from an exercise stress test).13 This parameter correlates strongly with BRS (r = 0.64; P = .001).13 We observed that symptomatic patients with LQT1 reduced their heart rate (HR) during the first minute of recovery significantly more than asymptomatic patients did, and those with marked HR reductions had a threefold greater risk of suffering cardiac events (odds ratio, 3.28; 95% confidence interval, 1.3 to 8.3; P = .012). In contrast, the phenomenon so clearly evident among patients with LQT1 is totally absent among patients with LQT2 and LQT3; this difference was expected, given that patients with LQT2 and LQT3 have a normal IKs current and are therefore less prone to the onset of life-threatening arrhythmias related to rapid changes in heart rate, especially increases. LQTS is regarded as a purely electrical disease without any mechanical alteration. Contrary to this view, a case-control study demonstrated the frequent presence of highly unusual echocardiographic abnormalities.14 These abnormalities include an increased rate of thickening in the early phase of contraction and the presence of a slow movement in the late thickening phase with a plateau morphology, sometimes accompanied by a second peak. They are more frequent in symptomatic than in asymptomatic patients, thus suggesting that they reflect the presence of an arrhythmogenic mechanism. Therefore, LQTS is not devoid of a mechanical component, although it is likely to be the consequence of abnormal electrical as indicated by the subsequent evidence that the calcium-entry blocker verapamil completely normalizes the contraction pattern,14 suggesting that symptomatic patients with LQTS have an abnormal increase in the intracellular calcium concentration before relaxation has completed and that this likely relates to an early afterdepolarization. The contraction abnormality would be the mechanical equivalent of an EAD. These findings have been recently confirmed in a large Norwegian study15 and provide definitive evidence that the electrical abnormalities characteristic of LQTS have the potential to produce highly specific mechanical alterations.14 The J-LN syndrome,16 characterized by congenital deafness, is due to the presence of two homozygous or compound heterozygous mutations on either the KCNQ1 or KCNE1 genes.2 Data on 187 patients with J-LN16 have shown clear differences versus the other types of LQTS, including LQT1, which shares with J-LN an impairment in the IKs current. J-LN is the most severe variant of LQTS. Almost 90% of the patients have cardiac events, 50% become symptomatic by 3 years of age, their average QTc is markedly prolonged (557 ± 65 ms), and they become symptomatic much earlier than any other major genetic subgroup of LQTS (Figure 93-3, A). For patients with J-LN, it has been possible to identify subgroups at lower risk, namely those with a QTc < 500 ms and those without syncope in the first year of life. Although the clinical diagnosis of J-LN is rather straightforward, it is important to genotype all these patients because the smaller group with KCNE1 mutations has a markedly less severe clinical course than that with mutations on KCNQ1 (see Figure 93-3, B). Figure 93-3 A, Kaplan-Meier curves of event-free survival comparing Jervell and Lange-Nielsen (J-LN) patients versus LQT1, LQT2, and LQT3 symptomatic patients. B, Kaplan-Meier curve of event-free survival in patients with J-LN syndrome and mutations in the KCNQ1 or KCNE1 genes. (A, Modified from Schwartz PJ, Spazzolini C, Crotti L, et al: The Jervell and Lange-Nielsen syndrome. Natural history, molecular basis, and clinical outcome, Circulation 113:783–790, 2006. B, From Schwartz PJ, Priori SG, Spazzolini C, et al: Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103:89–95, 2001.) The therapeutic approach to J-LN is made complex by the early age at which most of them become symptomatic and especially by the fact that β-blockers appear to have limited efficacy. Left cardiac sympathetic denervation may be less effective than in other patients with LQTS. Therefore, for many patients with J-LN an implantable cardioverter defibrillator (ICD) should be seriously considered, in addition to the traditional therapies. For the subgroups at lower risk,16 it may be reasonable to postpone a decision about ICD implant until 8 to 10 years of age. One special group of LQTS patients, which represents a tragic emotional burden for the parents and also for the responsible physicians, is constituted by infants and very young children presenting with recurrent cardiac arrest. These arrhythmias are poorly responsive to therapy and almost always require and ICD implant. Not uncommonly these infants are genotype-negative and their parents are unaffected. In 4 of these infants we have identified, by the use of exome sequencing in parents-child trios, de novo mutations in either CALM1 or CALM2, 2 of the 3 human genes encoding calmodulin. All mutation carriers were infants who exhibited recurrent ventricular fibrillation usually triggered by sympathetic activation, major QT prologation (QT <600 ms), intermittent 2:1 atrioventricular block, and presence of T wave alternans (Figure 93-2).17 Mutations altered residues in or adjacent to critical calcium binding loops in the calmodulin carboxyl-terminal domain and exhibited several-fold reductions in calcium binding affinity. Calmodulin mutations may contribute to unexplained sudden death during early deveopment. Given the characteristic features of LQTS, the typical cases present no diagnostic difficulty for physicians aware of the disease. However, borderline cases are more complex and require the evaluation of multiple variables besides clinical history and the ECG. Diagnostic criteria were proposed in 1985 with multiple updates.18 The last one has included the evaluation of the QTc in the fourth minute of recovery from an exercise stress test, given the reproducible finding that in LQT1 and LQT2 patients a QTc > 480 ms at this point was associated with 100% specificity.18 The new diagnostic criteria are listed in Table 93-2. The point score is divided arbitrarily into three probability categories: (1) ≤1 point = low probability of LQTS; (2) >1 to 3 points = intermediate probability of LQTS; and (3) ≥3.5 points = high probability of LQTS. Whenever a patient receives a score of 2 to 3 points, serial ECGs and especially several 24-hour Holter recordings should be obtained, because the QTc value in patients with LQTS can vary from time to time and from day to night. In this group with intermediate probability of LQTS, the presence of additional morphologic abnormalities can help with diagnostic decisions. Precordial leads are frequently more informative. Symptoms often appear in the first few years of life and can resemble epileptic convulsions.2 Table 93-2 Long QT Syndrome Diagnostic Criteria, 1993–2011 *QTc calculated by Bazett’s formula where QTc = QT / √RR. ‡Resting heart rate below the second percentile for age. §The same family member cannot be counted in A and B. From Schwartz PJ, Crotti L: QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 124:2181–2184, 2011. These diagnostic criteria were conceived in the premolecular era and should be used with common sense. Obviously, they cannot be of value in identifying the so-called silent mutation carriers (i.e., individuals with a disease-causing mutation and a QTc <440 ms), whose prevalence increases from 10% for LQT3 to 37% for LQT1.6 Molecular screening is essential for these subjects. The main value of the so-called Schwartz criteria is during a first contact with a patient and in clinical studies when uniformity in diagnosis is essential.
Long and Short QT Syndromes
Long QT Syndrome
Molecular Genetics of Long QT Syndrome
SCN5A (LQT3)
CACNA1c (LQT8): Timothy Syndrome
Prevalence
Romano-Ward Syndrome: Clinical Presentation
Cardiac Events and Their Relation to Genotype
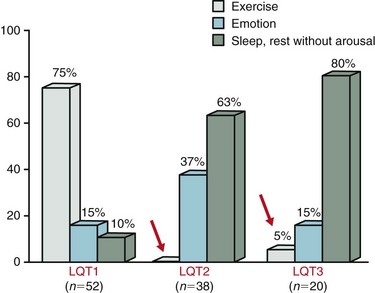
QT Interval Duration
T wave morphology
T Wave Alternans
Heart Rate and Its Reflex Control
Echocardiographic Abnormalities
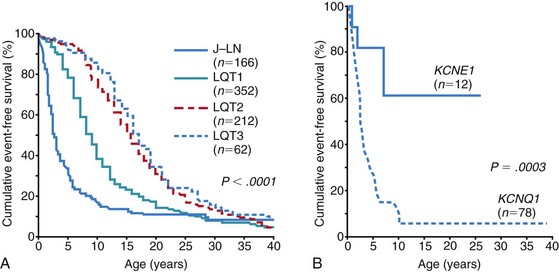
Jervell and Lange-Nielsen Syndrome: Clinical Presentation
Malignant Perinatal Long QT Syndrome: Clinical Presentation
Clinical Diagnosis
Long and Short QT Syndromes