Lipid Disorders
Mohamad Alkhouli, MD
Harish Jarrett, MBChB
Sara Sirna, MD
 ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
![]() Elevated plasma levels of low-density lipoprotein cholesterol, non–high-density lipoprotein cholesterol, lipoprotein(a), and apolipoprotein (apo) B-100.
Elevated plasma levels of low-density lipoprotein cholesterol, non–high-density lipoprotein cholesterol, lipoprotein(a), and apolipoprotein (apo) B-100.
![]() Reduced plasma levels of high-density lipoprotein and apo A-I.
Reduced plasma levels of high-density lipoprotein and apo A-I.
![]() Elevated plasma levels of triglycerides.
Elevated plasma levels of triglycerides.
![]() Skin xanthomas.
Skin xanthomas.
 General Considerations
General Considerations
Multiple epidemiologic studies have demonstrated the relationship between cardiovascular mortality and elevated plasma cholesterol levels. Lipid-lowering therapy, particularly with hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins), has demonstrated survival benefit in both primary prevention (patients without evidence of atherosclerotic cardiovascular disease [ASCVD]) and secondary prevention (patients with evidence of ASCVD). As a result, screening, risk stratifying, and treating dyslipidemia have become integral parts of preventive cardiovascular medicine.
A. Lipoproteins and Apolipoproteins
Cholesterol, cholesteryl esters, and triglycerides are the major lipids found in plasma. Cholesterol is an integral component of the cell membrane. It also serves a role in steroid hormone and bile acid synthesis. Triglycerides consist of fatty acid chains and phospholipids. Fatty acid chains are a primary source of energy in humans; phospholipids are key elements of all cell membranes. Cholesterol and triglycerides are insoluble in water and are transported in plasma by lipoproteins, classified as high-density lipoprotein (HDL) cholesterol, low-density lipoprotein (LDL) cholesterol, very-low-density lipoprotein (VLDL) cholesterol, intermediate-density lipo-protein (IDL) cholesterol, and chylomicrons. Lipoproteins consist of a lipid core and a water-soluble phospholipid outer layer that carry apolipoproteins, which are specific proteins that serve as coenzymes or receptor ligands or act as regulators of lipoprotein metabolism. Apolipoprotein (apo) A-I is present in HDL and promotes cholesterol efflux from tissues. Apo B, present as either apo B-100 (VLDL, IDL, LDL) or apo B-48 (chylomicron), serves as the LDL receptor ligand. Apo C-I, apo C-II, and apo C-III participate in triglyceride metabolism, and apo E is present on chylomicrons.
Lipoprotein(a) [Lp(a)] highly correlates with cardiovascular disease (CVD) and consists of an LDL particle and a specific apolipoprotein A. The structure of Lp(a) is similar to plasminogen and is thought to increase thrombogenesis. It is known to predict early atherosclerotic risk independent of other risk factors. Figure 1–1 illustrates the classes of lipo-proteins and their characteristics.
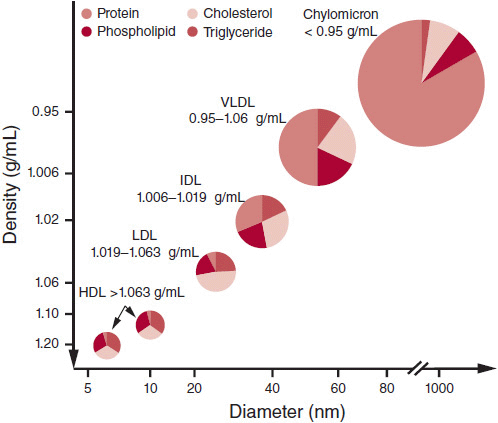
![]() Figure 1–1. Lipoprotein characteristics. HDL, high-density lipoproteins; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; VLDL, very-low-density lipoproteins.
Figure 1–1. Lipoprotein characteristics. HDL, high-density lipoproteins; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; VLDL, very-low-density lipoproteins.
B. Metabolism
Transportation of dietary lipids to peripheral tissues and the liver is known as the exogenous metabolic pathway. Here, triglycerides are hydrolyzed by pancreatic lipases and are emulsified by bile acids, leading to micelle formation. Micelle uptake occurs in the intestinal brush border where fatty acids are re-esterified and packaged together with apo B-48, phospholipids, and cholesterol to form chylomicrons. Chylomicrons then reach the portal circulation where they are hydrolyzed to release fatty acids by lipoprotein lipase (LPL). LPL activity is regulated through apo C-II, which activates LPL, and apo C-III, which inhibits LPL. The free fatty acids are used by muscle or stored as fat in an insulin-regulated process. As the particles get smaller, the cholesterol and apolipoproteins are transferred to HDL, creating a chylomicron remnant that is then taken up by the liver for degradation in an apo E–mediated process.
The endogenous metabolic pathway refers to transport of hepatic lipoproteins to peripheral tissues via LDL. It ensures a steady supply of substrate given that food supply varies. Here VLDL, a triglyceride-rich lipoprotein, serves a similar role to the chylomicron in the exogenous pathway. VLDL is derived from esterified long-chain fatty acids in the liver. Its triglyceride component is also hydrolyzed by LPL. VLDL acquires apolipoproteins (B-100, C-I, C-II, C-III, and E) from HDL and also exchanges triglycerides for cholesteryl esters in a process mediated by cholesteryl ester transfer protein (CETP). This mechanism allows cholesterol transfer from HDL to VLDL, allowing reverse cholesterol transport. Following hydrolysis, up to 40% of the resulting VLDL remnant, known as IDL, is taken up by the liver in an apo E–mediated transfer, and the remainder is converted to LDL by hepatic lipase. LDL particles mostly contain cholesteryl esters together with apo B-100 and are the main carriers of cholesterol. LDL is internalized through apo B-100, which binds to LDL receptors present on the cell surface. Cells are also capable of synthesizing cholesterol through an enzymatic process that involves HMG-CoA reductase. Figure 1–2 illustrates the endogenous and exogenous lipid metabolism pathways.
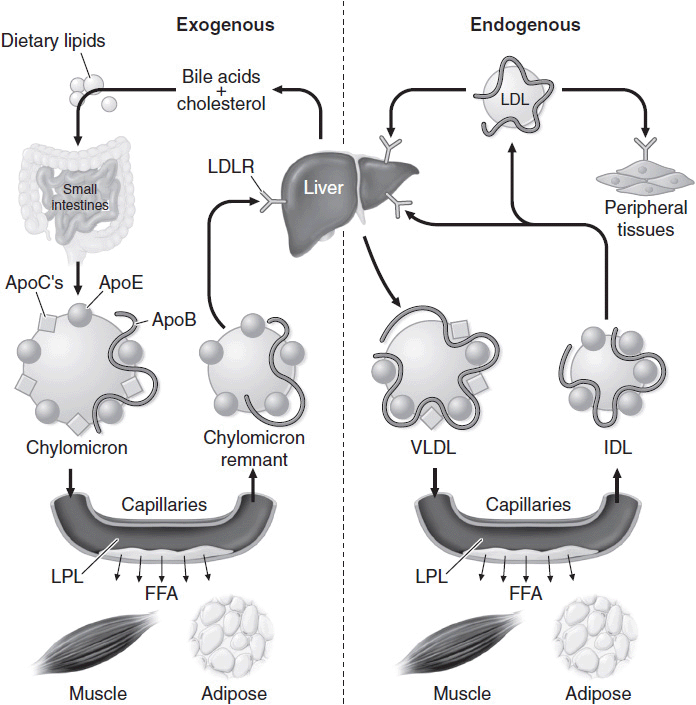
![]() Figure 1–2. Endogenous and exogenous pathways of lipid metabolism. FFA, free fatty acids; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LDLR, low-density lipoprotein receptor; LPL, lipoprotein lipase; VLDL, very-low-density lipoproteins. (Reproduced with permission from Longo DL, et al. Harrison’s Principles of Internal Medicine, 18th ed. New York: McGraw-Hill; 2012.)
Figure 1–2. Endogenous and exogenous pathways of lipid metabolism. FFA, free fatty acids; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LDLR, low-density lipoprotein receptor; LPL, lipoprotein lipase; VLDL, very-low-density lipoproteins. (Reproduced with permission from Longo DL, et al. Harrison’s Principles of Internal Medicine, 18th ed. New York: McGraw-Hill; 2012.)
There is an inverse relationship between HDL levels and atherosclerotic coronary artery disease (CAD). HDL is responsible for reverse cholesterol transport. It inhibits lipoprotein oxidation and maintains endothelial integrity. Synthesized mostly in the liver but also in the intestine, the nascent HDL molecule consists of apo A-I, phospholipids, and cholesterol. HDL acquires cholesterol in an apo A-I–mediated process, which is then esterified by lecithincholesterol acyltransferase (LCAT) within the HDL particle forming mature HDL-containing cholesteryl esters, which can be taken up by the liver. Cholesterol and triglycerides can also be transferred from HDL to VLDL and chylomicrons via CETP, a process that allows HDL to transport cholesterol between cells, VLDL and chylomicrons, and the liver.
C. Atherosclerosis
Dyslipidemia is a major predisposing factor in the development of atherosclerosis and ASCVD. Elevated plasma LDL concentrations can directly and indirectly contribute to atherogenesis. Oxidized LDL accumulates in subintimal macrophages through active scavenger receptor–mediated uptake that leads to cellular dysfunction, apoptosis, and necrosis. Unlike the LDL receptor–mediated endocytosis, this process is not regulated by cholesterol content. These macrophages are termed foam cells and form subintimal fatty streaks. This culminates in the release of proinflammatory and prothrombotic molecules, which causes endothelial injury. Oxidized LDL can also directly cause endothelial injury through interaction with the cell surface. Endothelial injury results in platelet aggregation and smooth muscle proliferation, which initiates atherosclerotic plaque formation. HDL serves a protective role in atherogenesis. Its antiatherogenic effects are exerted through maintenance of endothelial function, apo A-I–mediated reverse transport of macrophage cholesterol, antioxidant effects, and antithrombotic effects.
 Clinical Findings
Clinical Findings
Patients with dyslipidemia are asymptomatic with the exception of those who present with acute pancreatitis in the setting of severe hypertriglyceridemia. The clinical findings depend on the cause of dyslipidemia.
The most severe forms of hyperlipidemia are caused by genetic disorders of lipoprotein metabolism such as familial hypercholesterolemia (FH). FH is an autosomal dominant disease with defects in the gene for the LDL receptor. The mutant gene prevents the LDL receptor from removing LDLs from the blood. LDL cholesterol levels are typically in excess of 300 mg/dL. Common manifestations include fatty skin deposits called xanthomas over parts of the hands, elbows, knees, and ankles and around the corner of the eye (xanthelasmas). It frequently presents as premature CAD with chest pain (angina) or acute myocardial infarction. Other common primary causes of dyslipidemia are familial combined hyperlipidemia (both high LDL and triglyceride levels) and pure hypertriglyceridemia (very high triglyceride levels).
Table 1–1 summarizes the most common primary causes of dyslipidemia.
Table 1–1. Primary Gene Defects
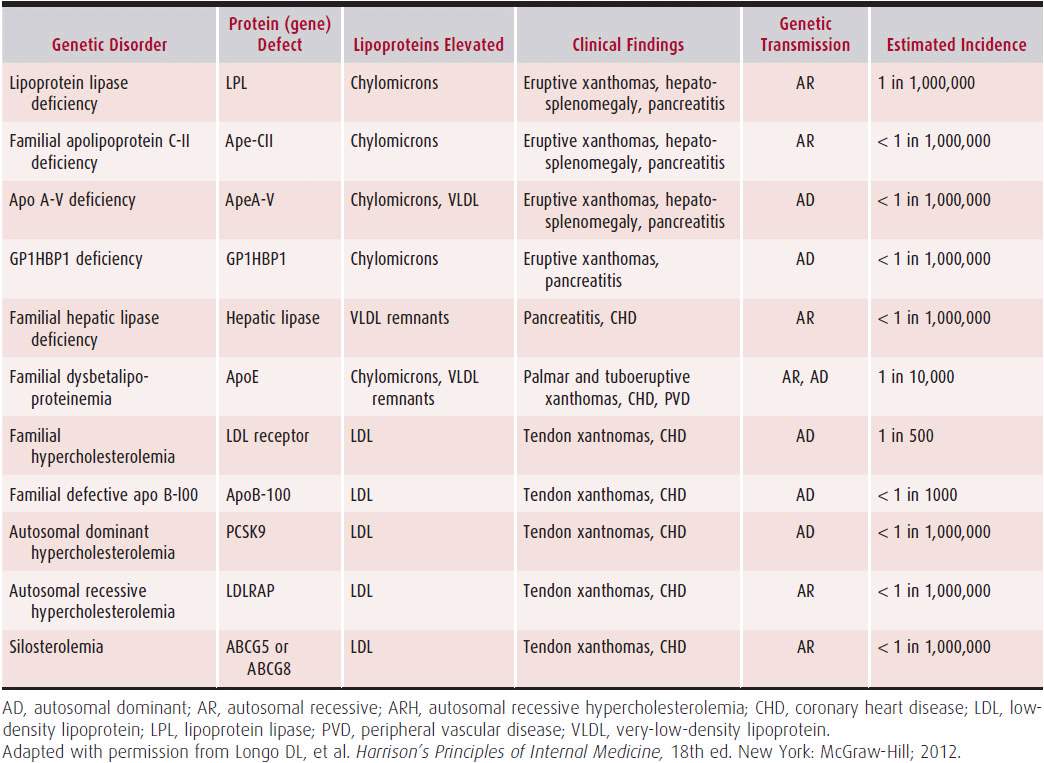
A. Secondary Causes of Dyslipidemia
Dyslipidemia develops in many patients as a result of an underlying metabolic abnormality rather than a primary dyslipidemia. Metabolic syndrome is characterized by abdominal obesity, hypertension, insulin resistance (± glucose intolerance), prothrombotic state, elevated triglycerides, small LDL particles, and low HDL levels.
Obesity affects over one-third of adults in the United States and represents a significant secondary cause of dyslipidemia due to caloric excess mostly derived from saturated fat and sugar. Decreased insulin sensitivity and increased carbohydrate intake cause an increase in triglyceride formation, which then enters the circulation in the form of VLDL. HDL levels are decreased in obesity. Similarly, patients with type 2 diabetes mellitus are dyslipidemic due to insulin resistance and subsequent insulin excess. This causes an increase in free fatty acid formation in the periphery and in the liver that translates to an increase in VLDL production and hypertriglyceridemia. Lipoprotein catabolism is down-regulated due to decreased LPL activity, resulting in further increases in plasma VLDL.
Hypothyroidism commonly results in dyslipidemia due to a decrease in hepatic LDL receptor activity and consequent delayed clearance of LDL. The severity of the dyslipidemia directly correlates with the degree of hypothyroidism, and correction reverses the dyslipidemia.
Liver disease can affect plasma lipid concentrations. Hepatitis is associated with increased VLDL production, and not surprisingly, severe disease and liver failure result in low plasma lipoprotein levels as synthetic function progressively decreases. Cholestatic liver disease results in significant hypercholesterolemia through the accumulation of lipoprotein-X, the deposition of which leads to the appearance of xanthomata in the skin.
Chronic kidney disease is associated with elevation in VLDL as a result of decreased lipolysis and clearance. Nephrotic syndrome results in marked hypercholesterolemia as a result of increased hepatic production of LDL and decreased clearance of VLDL, induced due to low plasma oncotic pressure.
Other secondary causes to consider include cigarette smoking, which decreases plasma HDL concentration. Commonly used drugs such as thiazide diuretics, β-blockers, and estrogen compounds can affect plasma lipid concentrations. Antiretroviral agents, particularly protease inhibitors, are associated with a lipodystrophy syndrome. Antipsychotics are also potential causative agents that are associated with weight gain and insulin resistance.
 Diagnosis
Diagnosis
The National Cholesterol Education Program, Adult Treatment Panel (NCEP-ATP) III recommends screening for lipid disorders with a fasting lipid profile every 5 years for adults over 20 years old. The US Preventive Services Task Force (USPSTF) advocates for routine screening of men and women above 35 and 45 years old, respectively. The USPSTF also recommends screening at earlier age if risk factors for coronary heart disease (CHD) are present. When lipid abnormalities are discovered, a thorough history should be performed, assessing for symptomatic CVD, as well as attempts to identify potential secondary causes of dyslipidemia. A thorough family history should be obtained, and risk factors such as sedentary lifestyle, obesity, poor diet, and cigarette smoking should be addressed. Physical examination may reveal xanthomas and xanthelasmas, which may aid in the diagnosis of a primary dyslipidemia. Basic metabolic panel, creatinine, and thyroid and liver function tests should be obtained to exclude any secondary causes of hyperlipidemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


