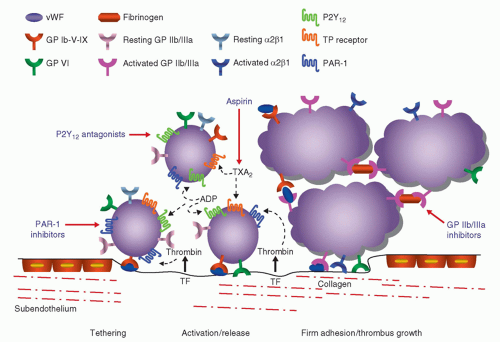
 FIGURE 3-1 Platelet mediated thrombosis. The interaction between glycoprotein Ib (GP Ib) and von Willebrand factor (vWF) mediates platelet tethering, enabling subsequent interaction between GP VI and collagen. This triggers the shift of integrins to a high-affinity state and the release of adenosine diphosphate (ADP) and thromboxane A2 (TXA2) which bind to the P2Y12 and TP receptors, respectively. Tissue factor (TF) locally triggers thrombin formation, which contributes to platelet activation via binding to the platelet protease activated receptor (PAR-1). (Adapted from: Angiolillo DJ et al. Circ J. 2010;74:597-607, with permission.) |
delays absorption and increases plasma levels, which require ˜3 to 4 hours. Since aspirin induces an irreversible COX-1 blockade, COX-mediated TXA2 synthesis is prevented for the entire life span of the platelet (˜7-10 days) (3).
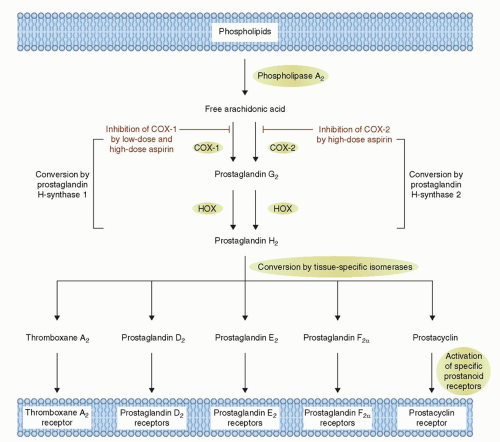 FIGURE 3-2 Mechanism of Action of Aspirin. Arachidonic acid, a 20-carbon fatty acid containing four double bonds, is released from membrane phospholipids by several forms of phospholipase A2, which are activated by diverse stimuli. Arachidonic acid is converted by cytosolic prostaglandin H synthases, which have both cyclooxygenase and hydroperoxidase (HOX) activity, to the unstable intermediates prostaglandin G2 and prostaglandin H2, respectively. The synthases are also termed cyclooxygenases and exist in two forms, cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2). Low-dose aspirin selectively inhibits COX-1, whereas high-dose aspirin inhibits both COX-1 and COX-2. Prostaglandin H2 is converted by tissue-specific isomerases to multiple prostanoids. These bioactive lipids activate specific cell-membrane receptors of the superfamily of G-protein-coupled receptors, such as the thromboxane receptor, the prostaglandin D2 receptors, the prostaglandin E2 receptors, the prostaglandin F2a receptors, and the prostacyclin receptor. (Adapted from: Patrono C. et al. N Engl J Med. 2005;353:2373-2383, with permission.) |
an irreversible effect on platelets, which lasts till their life span (7 to 10 days) (12).
TABLE 3-1 Large-Scale Randomized Clinical Trials Evaluating the Efficacy of Dual Antiplatelet Therapy with Aspirin and an Orally Administered P2Y12 Receptor Inhibitor in ACS/PCI Patients | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


