Chapter 7
Ischemia-Reperfusion
George P. Casale, Iraklis I. Pipinos
Based on a chapter in the seventh edition by Robert S. Crawford and Michael T. Watkins
The purpose of this chapter is to discuss the pathophysiology of severe acute ischemia, followed by acute reperfusion in the limbs and organs of the body. Ischemia-reperfusion (I/R) is a complex pathologic process involving intracellular and extracellular pathways that result in metabolic, thrombotic, and inflammatory changes in the affected tissues. A devastating component of I/R injury is the paradoxical increase in tissue damage associated with restitution of blood flow to ischemic tissues.
Haimovici1 described the development of myonephropathic syndrome in a few patients who underwent lower extremity revascularization after acute ischemia in the late 1950s. This report provided one of the first published clinical observations of limb I/R. These patients experienced ongoing lower extremity muscle necrosis and myoglobin-induced renal failure in the presence of palpable pulses. Cerra et al.2 completed an experimental animal and human clinical correlation of reperfusion in 1975. Histologic assessment of canine myocardium showed that restoration of blood flow was associated with the development of subendothelial hemorrhagic necrosis. Moreover, evaluation of the medical records and autopsy tissue from patients who died after aortic valve replacement provided evidence of subendothelial hemorrhagic necrosis in individuals subjected to more than 70 minutes of cardiopulmonary bypass. These findings implicated reperfusion injury as a cause of death in patients who underwent cardiopulmonary bypass for heart operations.
I/R injury has clinical relevance because it affects the outcome of patients after cardiac and vascular surgery, organ transplantation, plastic surgical reconstruction, and recovery from traumatic injury. The cellular damage that occurs as a consequence of I/R injury has been studied intensively in humans, experimental animals, and cell culture systems. To understand and characterize I/R injury from a biochemical perspective, it is useful to discuss this process in terms of its two components—ischemic injury and reperfusion injury.
Components of Ischemia-Reperfusion Injury
Injury During Ischemia
In the ischemic phase of I/R injury, the predominant mechanisms of injury result from tissue hypoxia or anoxia and stasis in the microcirculation (Fig. 7-1). The degree to which tissue from various organs is capable of tolerating ischemia varies widely and depends on each tissue’s baseline metabolic demand. Ischemia of human skeletal muscle under normothermic conditions is tolerated for more than 2 hours, whereas histologic evidence of ischemic injury develops in the jejunum after only 30 minutes of ischemia.3
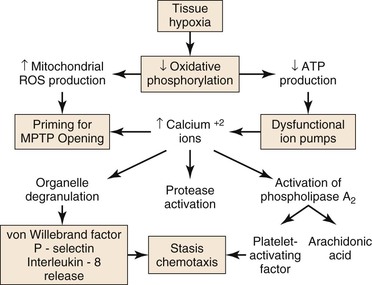
Figure 7-1 Injury during ischemia. Decreased oxygen supply activates a complex cascade of metabolic, inflammatory, and prothrombotic pathways and sets the conditions for MPTP opening during reperfusion. ATP, adenosine triphosphate; MPTP, mitochondrial membrane permeability transition pore; ROS, reactive oxygen species.
During ischemia, mitochondria deprived of O2 can no longer produce adenosine triphospate (ATP) by oxidative phosphorylation; consequently, cellular (ATP) falls rapidly with a concomitant increase in adenosine diphosphate (ADP), adenosine mononphosphate (AMP), and Pi. Glycolysis is stimulated, but is unable to produce sufficient ATP to meet the needs of the cell. The intracellular pH decreases with accumulation of lactic acid, progressively inhibiting glycolysis and activating the Na+/H+ antiporter that pumps H+ out of the cell. Because of ATP deficiency, the entering Na+ ions are not pumped out of the cell by Na+/K+ ATPase. High intracellular Na+ inhibits the Na+/Ca2+ antiporter, producing high intracellular Ca2+, which, in turn, activates degradative enzymes, including phospholipase A2, and proteases, such as calpains. In addition, ATP deficiency impairs ATP-dependent repair processes.4 Consequently, prolonged ischemia will lead to cell membrane damage and necrotic cell death.
As ischemia progresses, reactive oxygen species (ROS) are produced by and accumulate within the mitochondria.4,5 Increased mitochondrial matrix ROS together with increased cytosolic Ca2+, elevated Pi, and depletion of adenine nucleotides are most of the conditions needed for opening of the mitochondrial membrane permeability transition pore (MPTP), which causes necrotic cell death.6,7 The data indicate that the increase of mitochondrial ROS during ischemia primes the mitochondrial MPTP for opening, and that the magnitude of ROS damage to the mitochondria is the most important factor that determines the duration of opening of the MPTP, and therefore, the extent of tissue damage.4 Pore opening, however, is inhibited by the low intracellular pH during ischemia, but occurs during reperfusion when the pH increases, as is discussed in “Injury During Reperfusion” section in this chapter.
Tissue hypoxia leads to mobilization of neutrophils into the interstitium, where they have both beneficial and deleterious effects on tissue during reperfusion.8 Migration of neutrophils and macrophages to sites of inflammation is dependent on hypoxia-adaptive pathways.9,10 Activated neutrophils release the soluble mediators glutamate11 and adenine nucleotides (in the form of ATP or AMP)12,13 during ischemia that are converted to adenosine at the vascular endothelial surface. Adenosine protects the function of the microvascular endothelial barrier by re-establishing endothelial cell-cell contact after neutrophil transmigration. Transcellular metabolism (neutrophils provide ATP as a substrate for enzymes located on the endothelial membrane) and signaling are enhanced by hypoxia-induced transcriptional increases in functional endothelial surface apyrase (CD39), 5′-ectonucleotidase (CD73), and adenosine receptors (AdoRA2B).13,14 Polymorphonuclear neutrophils have a deleterious effect on local tissue by releasing factors that can disrupt the endothelial barrier. Activation of neutrophils by β2 integrins stimulates neutrophils to release soluble factors that induce endothelial cytoskeletal rearrangement, gap formation, and increased permeability. One neutrophil-derived permeabilizing factor is heparin-binding protein (HBP), also known as azurocidin or CAP37. HBP induces Ca2+-dependent cytoskeletal changes in endothelial cells and triggers macromolecular leakage in vivo.15
Injury During Reperfusion
Reperfusion injury represents the complex response to tissue injury when blood flow is restored after ischemia (Fig. 7-2). There are metabolic, thrombotic, and inflammatory components of reperfusion injury. The degree to which reperfusion either restores tissue integrity or exacerbates ischemic injury depends primarily on the duration of the ischemia. It is paradoxical that moderate ischemia followed by reperfusion may cause more fulminant postischemic tissue injury than seen with ischemia alone. However, without reperfusion, the loss of function in the brain, gut, heart, or limb may have a more catastrophic outcome than if perfusion is not restored.
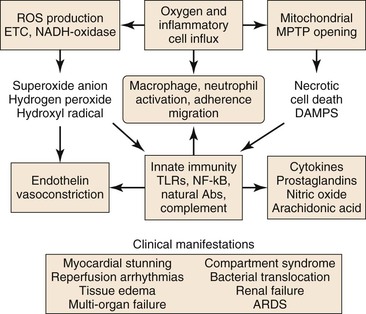
Figure 7-2 Injury during reperfusion. The return of oxygen, influx of inflammatory cells, and washout of metabolites contribute to cell death and an inflammatory, prothrombotic milieu that exacerbates tissue injury. Abs, Antibodies; ARDS, acute respiratory distress syndrome; DAMPS, danger-associated molecular patterns; METC, mitochondrial electron transport chain; MPTP, mitochondrial membrane permeability transition pore; NADH, reduced nicotinamide adenine dinucleotide; NF-κB, nuclear factor kappa B; ROS, reactive oxygen species; TLRs, toll-like receptors.
Reperfusion is characterized by a burst of ROS production (primarily by the mitochondrial electron transport chain), increased Ca2+ uptake into the mitochondrial matrix, and a shift of the intracellular pH from acidic toward neutral.6 Mitochondrial oxidative damage leading to programmed cell death is the major cause of tissue loss produced by reperfusion.16 The two forms of programmed cell death during reperfusion are apoptosis and necrosis.
Increased ROS generated during both ischemia and reperfusion can initiate apoptosis and necrosis, cell death programs mediated through the mitochondria. The BCL-2 family of proteins is central to regulating mitochondrial apoptosis. Antiapoptotic members, including BCL-2 and BCL-XL, act by blocking proapoptotic members, including BAX and BAK. The latter initiate apoptosis by increasing the permeability of the outer mitochondrial membrane, causing the release of proapoptotic proteins, such as cytochrome c.17 BH3-only proteins, proapoptotic BCL-2 family members, can initiate apoptosis by directly activating BAX and BAK or by blocking BCL-2 and BCL-XL.17 ROS initiate mitochondrial apoptosis by activating BH3-only proteins or by triggering opening of the mitochondrial MPTP, causing release of cytochrome c.7 Alternatively, apoptosis may occur via ROS-dependent lipid peroxidation of cardiolipin, a mitochondria-specific phospholipid of the inner membrane, which binds strongly to cytochrome c. The latter dissociates from peroxidized cardiolipin and is released from the inner membrane.18 The evidence, however, suggests that ROS-dependent necrosis, rather than apoptosis, is the main mechanism of cell death caused by I/R injury.6,7,19
Programmed necrotic cell death may be mediated by signaling via a tumor necrosis factor (TNF) death receptor20–22 or by direct opening of the mitochondrial MPTP.16,17 Both processes require calcium and oxidative stress.16,17 TNF receptor 1 (TNFR1) signaling for necrotic cell death begins with a conformational change caused by binding of TNF to the receptor. This conformational change promotes binding of cytosolic TNF receptor-associated death domain (TRADD) to a cytosolic domain of the receptor, initiating formation of a complex consisting of receptor-interacting protein 1 (RIP1) kinase, TNF receptor-associated factor 2/5 (TRAF2/5), cellular inhibitor of apoptosis 1 (cIAP1) and cellular inhibitor of apoptosis 2 (cIAP2). The TNFR1 complex is internalized and produces secondary cytosolic complexes, including the necrosome. The latter comprises Fas-associated death domain (FADD) protein, inactivated caspase 8, RIP1, and receptor-interacting protein 3 (RIP3). Activation of necrotic signaling requires phosphorylation of RIP1 and RIP3 and is critically dependent on ROS production by the mitochondrial electron transport system.21 The ultimate effector in this pathway is not yet established; however, the present data suggest that signaling leads to opening of the mitochondrial MPTP.20
Direct opening of the mitochondrial MPTP is central to tissue injury caused by ischemia and reperfusion.16,17,19 The mitochondrial MPTP is a nonselective protein channel spanning the inner and outer mitochondrial membranes at points of contact. When open, the pore permits free passage of water and solutes less than 1.5 kDa, causing osmotic damage to the mitochondria and uncoupling mitochondrial oxidative phosphorylation, the major source of cellular ATP, by dissipation of the pH gradient and the electrical potential across the inner mitochondrial membrane.4,6 Mitochondrial damage and collapse of oxidative phosphorylation promote disruption of metabolism and ionic homeostasis initiated before pore opening during ischemia, and thereby, promote and expand necrotic cell death. The composition of mitochondrial MPTP is not yet solved, but the voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT), and mitochondrial phosphate carrier (PiC) appear to be constituents.6 Cyclophilin D (CyP-D), a prolyl isomerase in the mitochondrial matrix, is an essential component of the mitochondrial MPTP and the central regulator of pore opening.16 Pore opening depends on mitochondrial calcium concentration (Ca2+ m) but is relatively insensitive to Ca2+ m per se, occurring at very high Ca2+ m. The sensitivity of pore opening to Ca2+ m is greatly enhanced by oxidative stress caused by increased mitochondrial ROS production during ischemia and a surge of mitochondrial ROS production during reperfusion.6 Oxidative stress promotes binding of CyP-D to an ANT/PiC dimer, facilitating a Ca2+-dependent conformational change in these proteins and opening of the mitochondrial MPTP. The evidence supports activation of CyP-D via binding of p53, which accumulates in the mitochondrial matrix in response to oxidative stress.16 Low intracellular pH that occurs during ischemia inhibits pore opening; however, during reperfusion, when blood flow is restored, the intracellular pH increases, and the mitochondrial MPTP opens.6
Reperfusion of ischemic tissue produces a burst of ROS production by the mitochondrial ETC and by parenchymal and endothelial cell reduced nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase and xanthine oxidase, neutrophils and macrophages, and necrotic cell death with opening of the MPTP.23 These processes lead to release of normally sequestered cellular proteins and protein fragments in addition to extracellular and cell surface accumulation of oxidatively modified proteins and lipids. Some of these structures have molecular configurations recognized by Toll-like receptors (TLR) of the innate immune system as “danger-associated molecular patterns” (DAMPS),24 and a growing body of work supports TLR-mediated inflammation as central to I/R injury.25 Reperfusion-associated DAMPS include heat shock protein 60 (HSP60), a ligand for TLR4; high-mobility group protein B1 (HMGB1), a ligand for TLR2, 4, and 9; low-molecular-weight hyaluronic acid, a ligand for TLR2 and 4; the extra domain A of fibronectin (EDA), a ligand for TLR2 and TLR4; and cardiac myosin, a ligand for TLR2 and 8.26 Ligand binding to TLRs present on parenchymal cells, endothelial cells, and circulating leukocytes activates TLR signaling pathways that lead to apoptosis or to nuclear factor kappa B (NF-κB) signaling.26 Inflammation induced by TLR-mediated NF-κB signaling is a significant contributor to I/R injury.25 NF-κB is a transcription factor present in the cytosol in an inactive complex with inhibitor of NF-κB (I-κB). Signaling via TLRs leads to removal and proteolysis of I-κB, permitting NF-κB to translocate to the nucleus, where it initiates transcription of proinflammatory genes, including those that code for TNF-α, interleukin-1β (IL-1β), and IL-6, as well as intercellular adhesion molecule-1 (ICAM-1), all of which recruit neutrophils and macrophages to the damaged tissue.23,25,27 In addition, NF-κB signaling may be initiated by binding of TNF to the TNFR1 receptor.21
Activation of innate immunity in response to DAMPS, during reperfusion, is not limited to their recognition by TLRs, but also occurs in response to their recognition by natural antibodies.27 These antibodies belong predominantly to the immunoglobulin-M (IgM) class, are secreted by B-1 cells (a specialized subset of B-cells) and are germline encoded.24 Many of these antibodies recognize epitopes formed by oxidative modification of lipids and proteins (e.g., during reperfusion). Some recognize normally sequestered self-antigen, for example, non-muscle myosin heavy chain II released as a consequence of cell injury during both ischemia and reperfusion.28 Reperfusion elicits an inflammatory response dependent on activation of complement by natural IgM antibodies.28 Binding of IgM natural antibodies to target molecules on the cell surface or in the extracellular environment activates the complement system, causing production of cleavage products, including complement component C3b, which tags cells and other targets for removal by macrophages, and C5a, a fragment chemotactic for neutrophils.27 Alternatively, the complement system may be activated in the absence of antibody by mannose-binding lectin (MBL) bound to cell surface carbohydrates.29 Complement activation, detected as tissue-bound C3b and MBL, occurs during ischemia and increases during reperfusion.30
A variety of inflammatory mediators are delivered to reperfused tissue by blood-borne cells (macrophages, lymphocytes, neutrophils, mast cells, platelets).31,32 Noncellular elements, such as the complement system, ROS, nitric oxide (NO), and proinflammatory and anti-inflammatory cytokines are also believed to modulate the complex scenario of reperfusion injury. The presence of O2, washout of lactatic acid, development of tissue edema, and influx of numerous cell types during reperfusion produce a complex environment in which numerous mediators contribute to inflammation and tissue injury. The cellular and molecular components of the I/R response will be discussed individually.
Leukocytes
In contrast to neutrophil activation during ischemia (discussed previously), neutrophil activation during reperfusion is characterized by a logarithmic increase in neutrophil chemotaxis, neutrophil–endothelial cell adhesion, and transmigration. In contrast to the slow transendothelial migration of resident neutrophils in the static ischemic environment, during reperfusion, flow modulates the interaction between neutrophils and endothelium. Transendothelial migration during reperfusion occurs in sequential steps: rolling, adherence, and transendothelial migration.9 The first step is initiated by increases in the surface expression of endothelial P-selectin (CD62P); CD62P interacts with its leukocyte ligand P-selectin glycoprotein ligand 1 (PSGL-1), which results in leukocyte rolling. Firm neutrophil adherence is modulated by interaction of the β2 integrins CD11a/CD18 and CD11b/CD18 with endothelial ICAM-1. Transmigration of leukocytes is facilitated by platelet–endothelial cell adhesion molecule-1 (PECAM-1), which is constitutively expressed along endothelial cell junctions. Once inside the interstitium, activated leukocytes release toxic ROS, proteases, and elastases.31 These substances contribute to increased microvascular permeability, edema, thrombosis, and parenchymal cell death.32,33 Neutrophil accumulation in the extravascular compartment is facilitated by chemotactic agents, including complement components C3a and C5a, chemokines such as IL-8, DAMPs, and leukotriene B4 produced by arachidonic acid metabolism.34 The release of chemotactic agents creates a gradient that attracts neutrophils from the intravascular space toward the interstitium.34–36
Cytokines-Chemokines/Antagonists
Multiple growth factors and cytokines are present at sites of inflammation, and each can influence the nature of the inflammatory response.37 There is a constant interaction between proinflammatory and anti-inflammatory signals in the vascular wall (Box 7-1).
Liver and skeletal muscle I/R have been correlated with increased levels of local and systemic cytokines that are believed to modulate local38–40 and systemic injury.41,42 TNF41,43 and macrophage inflammatory protein-2 (MIP-2) levels are markedly increased in hind limbs exposed to acute limb I/R.43,44 Direct cytokine antagonists (receptor antagonists and anticytokine antibodies),45 metabolic rescue agents,43,46 and anti-inflammatory molecules47–51 have been shown to ameliorate skeletal muscle injury and decrease tissue levels of proinflammatory cytokines when administered to animals during limb I/R. Recent literature suggests that administration of an IL-1 receptor antagonist attenuates spinal cord I/R injury by reducing neuronal necrosis and apoptosis.52 Administration of this same IL-1 receptor antagonist has also been shown to ameliorate renal I/R injury in animal experiments.53 Because this receptor antagonist is clinically used to treat rheumatoid arthritis, it may soon be used in clinical trials of I/R injury in humans. A word of caution regarding inhibition of cytokines or their receptors comes from the work of Shireman et al,54 who showed that chronic deficiency of monocyte chemoattractant protein-1 resulted in a decrease in the inflammatory response, as expected, but also inhibited skeletal muscle regeneration after chronic limb ischemia.
Mitochondrial Injury
The mitochondria are a dynamically growing and shrinking network of double-membrane organelles that comprise the primary energy-generating system of eukaryotic cells. Energy is generated by coupling ATP production with oxidative transfer of electrons from reduced substrates to molecular O2 (i.e., oxidative phosphorylation).55 This process of energy conversion is mediated by the mitochondrial electron transport chain that consists of five multiunit protein complexes tightly configured within the mitochondrial inner membrane.56 The largest of these is Complex I, which consists of 46 protein subunits and 9 redox centers, 1 of which is flavin mononucleotide (FMN) located at the terminus of the hydrophilic arm extending into the mitochondrial matrix. High-energy electrons from reduced nicotinamide adenine dinucleotide (NADH) enter the ETC by reducing the FMN of Complex I. The electrons pass through Complex I to its coenzyme Q (CoQ) reductase site located in the inner membrane and where CoQ is reduced. The latter shuttles the electrons to Complex III, which couples oxidation of reduced CoQ with reduction of (passage of the electrons to) cytochrome c located in the intermembrane space. Finally, the electrons are passed to Complex IV, which couples oxidation of cytochrome c with reduction of molecular O2 to water. Complex II can also shuttle electrons to CoQ by coupling conversion of succinate to fumarate with reduction of flavin adenine dinucleotide (FAD), which is present at the mitochondrial matrix side of the complex. Importantly, passage of two electrons through Complexes I, III, and IV is coupled with transfer of protons from the mitochondrial matrix across the inner membrane to the intermembrane space (i.e., a total of eight protons per electron pair).56 This process generates a pH gradient and a membrane potential used by Complex V to generate ATP from ADP and Pi.
Approximately 90% of inhaled O2 is consumed by mitochondria and reduced to water to produce energy. Under optimal physiologic conditions, 2% to 4% of the O2 consumed by mitochondria can capture single electrons that leak along the complexes of the electron transport chain generating the ROS superoxide anion (O•−).57 Oxidative stress caused by excess accumulation of ROS is held in check by mitochondrial superoxide dismutase, which converts it to H2O2 (another ROS) and catalase, which converts the latter to H2O and O2.57 During tissue ischemia, when O2 tension in the mitochondria is reduced (hypoxia), ROS generation paradoxically is increased, overwhelming oxidative defense enzymes and causing an excessive accumulation of ROS (oxidative stress).5 This happens because under tissue hypoxia with reduced availability of O2 as the final electron acceptor of the ETC, the NADH/NAD+ ratio in the mitochondrial matrix increases, thereby saturating the FMN of Complex I with electrons. Saturated FMN transfers electrons to available O2 in the matrix, producing O•−.57 This FMN-dependent process is amplified by (1) the accumulation of reduced CoQ generated by both Complex I and II under the condition of low electron flow, and (2) the presence of a pH gradient across the inner membrane. Under these conditions, present during ischemia, electrons back up through Complex I to its FMN, where electrons are passed to O2, generating O•−.57 With the onset of reperfusion, O2 returns to meet the conditions of increased NADH/NAD+, accumulated reduced CoQ, electron-saturated FMN, and an incapacitated electron transport chain. The surge of O2 thus provides a sink for flow of electrons from reduced FMN leading to a burst of O•−. The duration of this burst, and therefore, the extent of acute reperfusion damage are dependent on the duration and extent of ROS accumulation in the preceding ischemic phase of I/R. The evidence supports Complex I as the main source of this reperfusion burst of ROS generation.4
Complement
The complement system has long been recognized as an important mediator in innate immune defense and inflammation.58 Complement can be activated by any of three pathways—the antibody-dependent classical pathway, the alternative pathway, or the MBL-associated serine protease pathway. The classical pathway is initiated when IgM or IgG antigen-antibody complexes bind to C1, the first component of complement. Activation of the alternative pathway is triggered by microbial surfaces and complex polysaccharides, and results in the generation of C3. The lectin pathway is initiated by binding of the MBL protein to mannose and glucosamine residues on bacterial cell walls. Activation of these proteins results in cleavage of complement factors 4 and 2, followed by subsequent activation of C3.
There is considerable evidence to support a role of the complement system in hind limb I/R injury. In a model of rodent hind limb I/R, there was direct correlation between increased complement deposition, tissue injury, and increased glycolysis.59 Complement receptor 2–deficient (CR2−/−) mice exhibit deficient IgM secretion and are protected against I/R injury by defective B cells.60 IgM-secreting B-cells clones were studied from CR2 knockout mice and wild-type mice. Only one clone was found to restore injury to previously protected mice, thus suggesting that a single clone of self-reactive IgM can initiate complement-dependent I/R injury.61 In a follow-up study, peptides that bound the IgM clone were found to block injury in wild-type mice, a novel observation providing a potential therapeutic venue.62 Complement inhibition has been demonstrated to improve skeletal muscle contractility after I/R.63 C5-deficient mice have been shown to exhibit decreased lung vascular permeability, lung myeloperoxidase levels, and serum alanine aminotransferase levels, along with less skeletal muscle injury after reperfusion.64 Because the complement system is well described, and numerous inhibitors are available for each pathway, a beneficial role for these inhibitors in the brain, intestine, liver, and myocardium has been described.58
Nitric Oxide
NO is known to regulate vascular tone and to have potent anti-inflammatory and antithrombotic properties at the endothelial surface. It is also known to have concentration-dependent toxic effects on tissue and organ systems.65,66 The primary physiologic effect of NO results from stimulation of the activity of guanylate cyclase, accumulation of intracellular cyclic guanosine monophosphate, and a consequent decrease in intracellular calcium that results in relaxation of smooth muscle, decreased cardiac contractility, and reduced platelet and inflammatory cell activation. NO is produced from L-arginine by oxidation of one or two equivalent guanido nitrogens by NO synthase (NOS) in a reaction that requires O2, and the cofactors NADPH and tetrahydrobiopterin. There are at least three distinct genes for NOS; neuronal NOS (nNOS or NOS-1) and endothelial NOS (eNOS or NOS-3) are dependent on micromolar concentrations of calcium for activity and are constitutively expressed. The inducible isoform, iNOS (or NOS-2), is calcium independent and widely expressed in a variety of cells after exposure to specific stimuli. iNOS is often referred to as the high-output source of NO because it can be strongly induced by proinflammatory stimuli. However, the enzyme does not produce NO at a rate substantially greater than does eNOS or nNOS; rather, more of the protein can be transiently induced and activated at normal levels of calcium.67,68
One of the first observations implicating NO as a marker of ischemic injury was made by Beckman.65 His laboratory went on to propose that the production of NO and O2 radicals by neurons when the ischemic or hypoxic brain is reperfused might contribute to cerebral injury. Ischemia is believed to depolarize neuronal membranes and cause synaptic discharge of the excitatory neurotransmitter glutamate, which, in turn, opens the voltage-dependent, N-methyl-d-aspartate–specific glutamate receptor/ionophore and allows calcium to accumulate in the neuron. Calcium then activates an O2-dependent nNOS that oxidizes arginine to produce NO when O2 is readmitted to the brain by reperfusion. NO reacts with the O2 radical superoxide (O•−), also produced by reperfusion, to form peroxynitrite (ONOO−). ONOO− has been shown to interact directly with proteins (i.e., the mitochondrial electron transport chain, prostaglandin synthase) and amino acids (oxidizes critical cysteine residues, leading to deactivation of proteins), and triggers membrane lipid peroxidation, thereby altering membrane permeability.69 ONOO− is a powerful oxidant capable of nitrating phenolic moieties, such as tyrosine or tyrosine residues in proteins, and increases after traumatic brain injury.70 This powerful molecule also activates mitochondrial-dependent apoptosis, poly (ADP-ribose) polymerase (PARP),69 and NF-κB.71 Alternatively, some in vitro studies have shown that ONOO− may inhibit the generation of NF-κB.72,73 ONOO− is believed to diffuse for several micrometers before decomposing to form the powerful and cytotoxic oxidants hydroxyl radical and nitrogen dioxide. Beckman’s hypothesis is consistent with available evidence on the protective action of glutamate antagonists and O2 radical scavengers in limiting cerebral infarction after focal ischemia.66 Furthermore, ONOO− decomposition catalysts are known to limit reperfusion injury in a variety of organ systems.74
There are also anti-inflammatory effects of NO that deserve mention. NO donors have been shown to ameliorate tissue injury in in vivo and ex vivo models of intestinal, neuronal, and hepatic I/R.75–77 NO is known to inhibit activation of NF-κB,78,79 which regulates a wide variety of proinflammatory cytokines and adhesion molecules. NO alone is an important antiatherosclerotic autacoid with antiaggregatory effects on platelets and with antioxidant, anti-inflammatory, and antiproliferative effects on the vasculature.80 NO inhibits vascular permeability in the postischemic intestinal microcirculation, probably by preventing endothelial cell contraction and gap formation.81 Endogenous NO also inhibits neutrophil adhesion to endothelium.82 Finally, NO regulates the activation of platelets by decreasing aggregation and adhesion responses.83 The duration of ischemia, magnitude of the injury, and specific organ bed will determine whether NO has a protective or deleterious effect. In contrast to the deleterious effects of NO when synthesized in large amounts by inflammatory cells via iNOS, the protective effects of NO appear to be related primarily to endogenous sources of constitutively synthesized NO generated by eNOS or nNOS or exogenous pharmacologic doses of NO donors.84
Endothelin
Endothelin (ET), a 21-amino acid peptide with potent vasoconstrictor properties,85 is produced mainly in endothelial cells, yet it is present throughout the entire cardiovascular system. Shear stress, growth factors, vasoactive hormones, and I/R are major stimuli for its release. Three distinct human ET-related genes have been cloned: “classic” ET-1, ET-2, and ET-3.86 ET-1 is the most predominant isoform in blood vessels and is synthesized by endothelial cells. ET-1 is produced as a precursor peptide that is cleaved by a converting enzyme into its active form. ET plays a major role in the regulation of vascular tone through its potent vasoconstrictive actions. ET acts on two different types of G-protein–coupled receptors: ET-A and ET-B. ET-1, the most potent vasoconstrictor, mediates its effects mainly through ET-A receptors present on vascular smooth muscle cells.87 ET-B receptors mediate both vasodilatory effects of ET through prostacyclin and NO (ET-B1) and vasoconstrictive effects on vascular smooth muscle (ET-B2).88,89
Increased production, upregulation of its receptors, enhanced sensitivity, and potentiation of the action of serotonin and norepinephrine all potentiate ET’s actions during I/R.90,91 Bosentan, a mixed ET-A/ET-B endothelin receptor antagonist, was shown to exert a marked tissue protective effect, as assessed by histologic evaluation in a rat model of myocardial I/R. This effect was probably the result of antioxidant protection derived from preservation of levels of the antioxidant enzymes superoxide dismutase and catalase in the treated animals.92 McMurdo et al93 showed that the vasoconstrictive effects of ET-1 in the coronary vasculature of the rabbit were primarily mediated by the ET-A receptor. However, FR139317, an ET-A receptor antagonist, failed to decrease infarct size after the induction of ischemia and reperfusion in that same model.93 A more contemporary assessment of the action of BQ-123, a selective ET-A receptor antagonist, revealed decreased myocardial levels of lipid peroxidation; an increase in NO, glutathione, catalase, and superoxide dismutase activity; and a decrease in the ratio of infarcted area to area at risk compared with I/R alone.94 Further evidence has proved that ET-A antagonism is useful in I/R. The ET-A antagonists ABT-627, S-0139, BSF208075, and LU135252 all show significant tissue protection in models of I/R.95–98
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


