Chapter 5
Atherosclerosis
Christopher D. Owens
Vascular surgeons care for a diverse set of clinical manifestations related to atherosclerosis—from symptomatic transient cerebral ischemic attacks to aortoiliac occlusions to aortic aneurysm repairs in patients with advanced coronary heart disease (CHD). However, despite the wide range of manifestations, culprit lesions are more alike than different. Accumulation of large amounts of cholesterol ester in the arterial wall and formation of complex advanced plaque are common to all these lesions. Although their formation insidiously spans decades, atherosclerotic lesions can reach a clinical horizon within minutes and manifest as catastrophic myocardial infarction (MI), stroke, or limb ischemia. Far from being a simple cholesterol deposition problem, plaque formation and evolution are now recognized as much more dynamic in nature. Rather than following a progressive degenerative process, individual plaque formations may progress and regress or undergo growth spurts. Inflammatory changes within the fibrous cap may render them vulnerable to rupture or erosion and ulceration. An understanding of each of these processes helps the vascular surgeon in developing evidence-based treatment strategies for each of the clinical syndromes of atherosclerosis, which together constitute a massive public health problem.
Atherosclerotic-related cardiovascular disease (CVD) is the leading cause of death in every region of the world except sub-Saharan Africa, and it has now eclipsed infectious disease in most underserved nations.1 However, during the last two decades, CHD age-specific death rates fell by greater than 40% in high-income countries.2 The prolonged survival has translated into growing disease prevalence and a staggering financial burden. It is estimated that 80,700,000 Americans have CVD with an estimated annual cost of treatment of $448.5 billion.3 Globally, 202 million people have peripheral arterial disease (PAD), which disproportionately affects blacks and individuals living in low to middle-income countries.1,3 Particularly troubling is the high prevalence of cardiovascular risk factors in children and young adults.4,5 A sedentary lifestyle, abdominal obesity, and poor diets contribute to dyslipidemia and high blood pressure. Autopsy studies in children and young adults demonstrate a link between these risk factors and early lesions.4
This chapter outlines the existing theories of the pathophysiology of atherosclerosis and the relationship to traditional and emerging risk factors. It also describes our current conceptual understanding of the fundamental biology of atherosclerotic plaque.
Normal Artery Structure and Function
A detailed presentation of normal arterial structure can be found elsewhere in this text (see Chapters 3 and 4); however, the relevant anatomy is presented here. Histologically, the arterial wall consists of three distinct layers: the intima (endothelium and subendothelial space), the media (smooth muscle cells [SMCs]), and the adventitia (vasa vasorum and connective tissue elements). The intima consists of the endothelium, its basement membrane, and the subendothelial space, which extends to the internal elastic laminae. The endothelium is a single layer of cells lining the inner surface of all blood vessels.
Endothelium
The endothelium is an organ with autocrine, paracrine, and endocrine functions that serve to regulate blood vessel tone and thrombogenicity. The endothelium synthesizes an array of factors that regulate vascular tone (e.g., nitric oxide [NO], hyperpolarizing factors, prostaglandins, endothelin, angiotensin II) and interactions with blood elements (e.g., adhesion molecules, thrombomodulin, plasminogen activators), which all work in balance to maintain vascular homeostasis.
Endothelial dysfunction is regarded as the earliest manifestation of vessel injury and is present before histologic evidence of atherosclerosis.6 Hence functional testing of the endothelium is an important tool for investigators studying atherosclerosis. Endothelial function can be detected in vivo by invasive and noninvasive techniques. Catheter-directed intraarterial administration of acetylcholine has been used to detect the presence of endothelium-dependent vasodilatation in the coronary and peripheral vasculature. Paradoxical contraction with low doses of acetylcholine is regarded as positive for endothelial dysfunction in the coronary circulation. An acute increase in blood flow can also stimulate endothelial relaxation. Flow-mediated, endothelium-dependent vasodilatation can be measured noninvasively by high-resolution ultrasound techniques.7 The primary mediator of flow mediated vasodilation is NO.8 Administration of nitroglycerin can detect the ability of vascular smooth muscle to relax in the presence of exogenous NO and thus detects endothelial-independent relaxation.
Subendothelial Space
The subendothelial space is particularly relevant in atherosclerosis because it is the location where atherogenic particles are retained and modified so that they can be taken up by macrophages and SMCs.9 In terms of volume, the major component of the extracellular intimal matrix is proteoglycans, which form a three-dimensional network that fills 60% of the space.9 The remaining mass of this compartment is filled with type IV collagen of the basement membrane, type I and III collagen randomly dispersed in this matrix, and occasional elastin fibers. Extending from the proteoglycans are sulfated glycosaminoglycan chains, which serve to interact with positive residues on apolipoproteins. Large versican-like proteoglycans containing chondroitin sulfate readily interacts with apolipoprotein B-100 (apoB-100), the apolipoprotein associated with low-density lipoprotein (LDL).10 The stoichiometry is such that 1 µg of versican can bind to 50 to 100 µg of LDL at physiologic ionic strength, thus suggesting that several lipoprotein particles are retained on each proteoglycan molecule.10,11
Smooth Muscle Cells
In children, very few SMCs or macrophages are present in the subendothelial space; however, with age and lesion formation, their concentration increases. In autopsy studies, abundant SMCs are found in the coronary arteries in atherosclerosis-prone areas, but they are relatively sparse in areas resistant to atherosclerosis.12 Whereas human medial SMCs predominantly express proteins involved in the contractile function of the cell, those found in the intima express lower levels of these proteins, have a higher proliferative index, and exhibit greater synthetic capacity for extracellular matrix, proteases, and inflammatory cytokines.13,14 These “synthetic” SMCs migrate more readily and can produce up to 25 to 46 times more collagen than “contractile” SMCs can. Thus SMCs directly contribute to intimal thickening as a result of increased proteoglycan production, inflammatory cell recruitment, and retention of atherogenic particles within the subendothelial space.
Vasa Vasorum
The vasa vasorum may contribute to atherosclerosis as well. First-order vasa vasorum run longitudinally to the lumen of the host vessel, whereas second-order vasa vasorum are arranged circumferentially. As plaque volume increases, there is an increase in the vasa vasorum visible on histologic section. Whether this is a response to plaque growth or contributes to plaque growth is unknown. Although the development of vasa vasorum in plaque involves angiogenesis, it is unclear what signals this process. Atherosclerotic vasa vasorum can proliferate and thereby lead to extensive neovascularization directed toward lipid-rich atheroma. The neovessels of plaque may provide an additional portal for trafficking leukocytes to promote the inflammatory process or become friable and contribute to intraplaque hemorrhage.15
Atherosclerotic Lesions
Lesions of the arterial wall have been divided into six types based on their histopathologic features.16,17 However, although useful for comparing pathologic specimens, the classification system has limited clinical practicality. The assumption of a relentless, linear progression of the disease has recently been challenged, and though this assumption is true under experimental conditions in some animal models,18,19 it is not necessarily the case in humans.20,21
Autopsy studies from the Bogalusa Heart Study and the Pathobiological Determinants of Atherosclerosis in Youth study demonstrate that atherosclerotic lesions form in early childhood and increase with age and that a significant percentage of them are raised.4,22–24 In an intravascular ultrasound (IVUS) study of heart donors, 17% of individuals younger than 20 years had evidence of atherosclerosis.25 Notably, both the Bogalusa Heart Study and the Pathobiological Determinants of Atherosclerosis in Youth study emphasize that the number and severity of early lesions are directly related to known cardiovascular risk factors, thus suggesting that a paradigm shift is needed to address risk factors in childhood rather than pharmacologic therapy later in the disease process.
Diffuse intimal thickening has been identified in the atherosclerotic-prone areas of coronary arteries as early as 36 weeks of gestation.26 Although the fatty streak—dominated by lipid-filled macrophages—is itself benign, it is the precursor of the more clinically relevant late lesion. The fatty streak is the first lesion visible to the naked eye, and because of its positive Sudan staining, it is referred to as sudanophilic. Its yellow color is attributed to lipid in the form of cholesterol and cholesterol esters within macrophages and SMCs. From the aforementioned studies, it is clear that atherosclerosis begins early in life in all races, is associated with risk factors, and occurs in anatomic locales susceptible to the formation of more advanced fibrous plaque.
Advanced lesions, or fibrous plaque, are characterized histologically by the amount of extracellular lipid and fibrous connective tissue seen. They are whitish in gross appearance and are elevated so that they protrude into the lumen. These fibroatheromas are prone to produce clinical sequelae by erosion of the surface endothelial cells, rupture of the fibrous cap, erosion of a calcium nodule, or intraplaque hemorrhage (Fig. 5-1).16,27 Plaques differ in consistency and may be relatively soft and friable or densely sclerotic and calcific. Likewise, some have well-formed fibrous caps, whereas others are covered by a narrow zone of loose connective tissue or by endothelium alone.
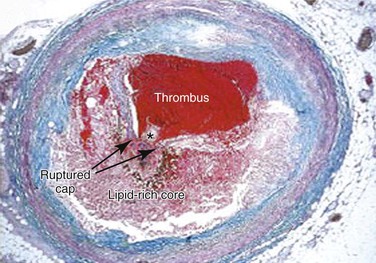
Figure 5-1 Thrombotic complication of a fibroatheroma seen on a trichrome-stained (thrombus and intraplaque hemorrhage stain red; collagen stains blue) cross-section of a human coronary artery. The fibrous cap has ruptured (area between the arrows), and the highly thrombotic lipid core is exposed to circulating blood with the subsequent production of acute occlusive thrombosis.
The necrotic core usually occupies the deeper central regions of the plaque and contains amorphous lipid and cholesterol crystals. The term atheroma is derived from the Greek word athere, meaning “porridge-like gruel.” SMCs and inflammatory cells are located adjacent to the necrotic core and at the shoulders of the plaque, where it is most susceptible to rupture.
The fibrous caps contain varied levels of SMCs adjacent to the collagen and basement membrane. These cells have reduced proliferative ability and may be thought of as senescent. However, the fibrous cap of ruptured plaque is often infiltrated with foam cells, which are largely of macrophage origin28 and thus indicative of active inflammation and vulnerability to rupture. A priori identification of these so-called thin-cap fibroatheromas is an active area of cardiovascular imaging research.29
Theories of Pathogenesis
Each of the well-regarded theories of atherosclerosis discussed in this section attempts to explain the underlying pathogenesis of atherosclerotic plaque. From the time initially proposed, each theory has undergone a steady evolution as our methodology of scientific inquiry has advanced from histologic descriptors to powerful molecular mechanisms. Each theory, held accountable over time, must incorporate new scientific data within a clinical context of risk factors and biomarkers to result in a model of atherosclerotic lesion formation.
Lipid Hypothesis
Cholesterol has been one of the most studied molecules in biomedical research. Since it was first isolated from gallstones in 1784, organic chemists and biochemists, physiologists and physicists, epidemiologists, and other medical scientists have devoted significant portions of their careers to its study and have collectively produced a body of work culminating in 13 Nobel Prizes.30 The modern era of cholesterol research began in St. Petersburg, Russia, at the turn of the 20th century, when Nikolai Anitschkow produced vascular lesions in rabbits by feeding them purified cholesterol dissolved in sunflower oil.31,32 These lesions closely resembled those seen in human atherosclerosis. He determined that the earliest lesions consisted of foam cells that were Sudan positive and contained birefringent crystals and hypothesized that they were caused by elevated serum cholesterol. He also noted a distinctive pattern consisting of location of the lesions near arterial branch points and concluded that this was probably determined by hemodynamic factors. Nevertheless, this work was criticized because the levels of cholesterol produced were too high and could not be experimentally reproduced in more conventional animal models such as rats and dogs.31,32
Cholesterol is absolutely insoluble in water, and the early work done in Russia provided no clues as to how it was transported to the arterial wall and formed lesions. The discovery that cholesterol was associated with proteins that allowed it to be transported in the aqueous environment led to investigations into lipoproteins. The first investigation in which the lipoprotein content in whole serum was accurately quantified involved analytic ultracentrifugation and was led by John Gofman of the Donner Laboratory at Berkley.33 Lipoprotein fractions were isolated and characterized by their densities and floatation characteristics.33 Of importance, this group noted that it was not simply the total cholesterol that was important but the species of lipoprotein contained within the cholesterol.34
Cholesterol is transported in the aqueous environment by esterification of the sterol of long-chain fatty acids and packaging of these esters with the hydrophobic cores of plasma lipoproteins. With its polar hydroxyl group esterified, cholesterol remains sequestered within this core, which is essentially an oil droplet composed of cholesteryl esters and triglycerides, solubilized by a surface monolayer of phospholipid and unesterified cholesterol and stabilized by protein. In persons who are fasting, lipids circulate in plasma as lipoprotein particles that are defined on the basis of their density as very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), LDL, and high-density lipoprotein (HDL). The protein of the VLDL, IDL, and LDL molecule is apoB-100, which has β-electrophoretic mobility.
The major milestone in the lipoprotein field was discovery of the defective gene associated with familial hypercholesterolemia by Goldstein and Brown. In fibroblasts cultured from normal human subjects, the activity of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-controlling enzyme in cholesterol biosynthesis, is regulated by the content of LDL (but not HDL) in the culture medium. Goldstein and Brown found that 3-hydroxy-3-methylglutaryl coenzyme A reductase was not sensitive to normal feedback regulation by LDL in cultured skin fibroblasts from homozygous familial hypercholesterolemia patients.35,36 They then determined that the defect was related to deficient binding of LDL to the cells of patients with familial hypercholesterolemia and that suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity was related to the amount of LDL bound.37 Therefore these patients had overproduction of cholesterol because they lacked the appropriate receptor. They demonstrated that cellular uptake of LDL absolutely requires the LDL receptor, without which the LDL cholesterol concentration builds up to 800 to 1000 mg/dL (20.72 to 25.90 mmol/L). Recognition of the importance of this work resulted in Goldstein and Brown sharing the Nobel Prize in Physiology and Medicine in 1985.
Epidemiologic evidence from numerous studies, including the Japanese migration studies38,39 and the Framingham Heart Study,40 demonstrated the association between cholesterol and incident CHD. Other studies showed that diets rich in unsaturated fats resulted in lowered cholesterol and reduced cardiovascular events.41
Widespread acceptance of the causative role of cholesterol began with publication of the Coronary Primary Prevention Trial, sponsored by the National Institutes of Health.42,43 This trial demonstrated that reducing cholesterol with the bile acid–binding resin cholestyramine would reduce cardiovascular events. A decade later, this trial was reinforced with powerful data emerging from the statin era.44 Although the primacy of the “cholesterol hypothesis” has been called into question, there is no doubt that lipids play a critical role in the pathogenesis of atherosclerosis.
Response-to-Injury Hypothesis
The basis of this hypothesis lies in the similarities between atherosclerosis and the response of arteries to experimental injury. Although the origins of the hypothesis date back to Virchow, the modern version of the hypothesis builds on earlier work by Duguid (1949),45 Poole and Florey (1958),46 and French (1966)47 and was formally advanced in 1973 by Ross and Glomset,48 who emphasized the importance of injury to the endothelium as a seminal event initiating the development of atherosclerosis lesions. Even though the definition of the term injury has been modified over the last three decades,49 all response-to-injury hypotheses have emphasized the primacy of the endothelium in stimulating the cascade of events leading to lesion formation.48–53
Endothelial injury may result from mechanical disruption, exposure to toxic or infectious agents, or endogenous inflammatory signals. Injury to the endothelium allows adhesion of platelets and an influx of LDL and other serum factors into the subendothelial space. Platelets release their alpha granules and stimulate migration of SMCs into the intima, where they proliferate and form a thickened neointima responsible for narrowing of the arterial lumen. Restoration of a healthy endothelial cell layer abates the process. During work on this hypothesis, Ross et al performed experiments leading to the discovery of platelet-derived growth factor (PDGF).54,55
Others counter that it is not injury to the endothelium that is the initiating event but rather retention of inflammatory lipids in the subendothelial space, which renders a particular area susceptible to atherosclerosis. By noting that LDL accumulates in the intima within 2 hours after a bolus infusion56 and that the accumulation occurs before the formation of fatty streaks,57,58 they concluded that retention of LDL is the initiating event of atherosclerosis. The so-called response-to-retention theory purports that retained apoB–containing lipoproteins stimulate a macrophage- and T cell–dominated inflammatory response in the arterial wall.59,60
Monoclonal Hypothesis
During the same year that Ross and Glomset were formulating the response-to-injury hypothesis of atherosclerosis, another group of investigators at the same institution (University of Washington) were writing their own hypothesis of atherosclerosis. This hypothesis suggests that each lesion of atherosclerosis is derived from a single SMC that serves as a precursor for the clonal expansion of proliferating SMCs.61 The hypothesis put forth by Benditt and Benditt uses the concept that in every female cell there is only one active X chromosome and the progeny of that cell will express the same X chromosome as the parent cell. Glucose-6-phosphate dehydrogenase (G6PD) has two isoforms that can be separated by electrophoresis. Its gene is located on the X chromosome and can therefore be used to identify the progeny of a parent cell. This approach was used to determine that uterine leiomyomas are composed of cells with the same active X chromosome, whereas adjacent normal myometrium was composed of a mixture of cells containing both G6PD isoforms and, therefore, that both X chromosomes were active.62
The Benditts examined a series of atherosclerotic plaque from four black females and compared them with adjacent normal areas of arterial wall. They determined that SMCs from the plaque contained only one G6PD isoform, whereas adjacent control areas contained a mixture of isoforms. This allowed them to conclude that each lesion is a clonal outgrowth derived from a single precursor SMC located in the intima. It is noteworthy that SMCs within individual human neonatal intimal thickenings are monoclonal in origin, whereas cells from the subjacent media are polyclonal.
Others challenged the hypothesis by noting that identification of a single enzyme phenotype does not necessarily imply clonal origin.63,64 Regardless, the work was importantly heuristic. It hinted toward the role that modern molecular biology would play in unraveling the genetic basis of atherosclerosis.
Atherosclerosis as a Chronic Inflammatory Disease
The original version of the response-to-injury hypothesis of atherosclerosis initially proposed that endothelial denudation was the first step in atherosclerosis.48 Subsequent versions of the hypothesis proposed that an endothelium chronically bathed in serum with high concentrations of LDL or exposed to other cardiovascular risk factors would render susceptible areas of the endothelium dysfunctional or activated.52 Indeed, an intact endothelium may be a necessary factor for lesion progression, and it is now clear that developing atheromas are covered by an intact endothelium throughout most stages of lesion progression.17,49,65–67 In humans, only the most advanced ulcerated lesions are focally devoid of endothelium. The injury results in increased adhesiveness and an increase in permeability of the endothelium to inflammatory cells (Fig. 5-2).
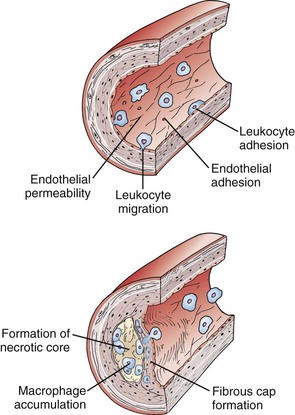
Figure 5-2 Initiation and progression of atherosclerotic plaque. Cardiovascular risk factors, hemodynamic forces, toxins, and infectious agents interact with the vessel at the level of the endothelium to produce injury, resulting in decreased nitric oxide (NO) production and increased permeability. Once injured, the endothelium increases the expression of leukocyte adhesion molecules such as vascular cell adhesion molecule-1, intracellular adhesion molecule-1, and P- and E-selectin, which increases the adherence of macrophages and other leukocytes. Permeability of the endothelium also increases and permits entry of leukocytes and lipoproteins into the subendothelial space. Chemokines and cytokines such as monocyte chemotactic protein-1 (MCP-1) and interleukin-8 (IL-8) further enhance the recruitment of leukocytes and smooth muscle cells (SMCs) into the subendothelial space. Lipoproteins retained in the subendothelial space are biochemically modified such that they can be taken up by macrophages and SMCs to form foam cells. Foam cells at the central-most position of the developing atheroma become necrotic and form the central lipid core, whereas the shoulder regions contain SMCs, macrophages, and other leukocytes. Platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) stimulate SMC migration and collagen formation in the subendothelial space, as well as formation of the fibrous cap.
Atherosclerosis is now recognized as an inflammatory disease, and components of the innate and adaptive immune system are involved in every step of the atherosclerotic process. Much of our modern understanding comes from examination of human pathology specimens and transgenic animals. Genetic deletion of apolipoprotein E (apoE−/−) or the LDL receptor (ldlr−/−), which produces mice with severe hypercholesterolemia and atherosclerotic lesions with features of mature human atheroma, has become a cornerstone in atherosclerosis research laboratories.68–70 The importance of the LDL receptor in cholesterol regulation was noted by Goldstein and Brown when studying patients with familial hypercholesterolemia. Apolipoprotein E suppresses atherosclerosis, and apoE−/−– mice have very low amounts of pre-β HDL and their plasma is poor at promoting the efflux of cholesterol from lipid-laden macrophages.71 Crossbreeding these mice with other strains carrying null mutations in immunologically relevant genes produces a robust research tool to dissect out the contribution of individual components of immune pathways in atherosclerosis. For example, some of the earliest approaches using compound mutant mice involved the global loss of the entire adaptive immune system. Rag1−/− or Rag2−/− mice lack the V(D)J recombinase required to form lymphocyte antigen receptor genes and hence have a complete loss of B and T cells. In apoE−/− mice, a regular chow diet produced plasma cholesterol levels between 390 and 470 mg/dL. Double knockout apoE−/−/Rag1−/− animals had a 40% reduction in aortic atherosclerotic lesion reduction compared with immunocompetent animals and provided an important piece of evidence of the role cellular immunity plays in the pathogenesis of atherosclerosis.72
LDL Retention
Trafficking of circulating LDL into and out of the subendothelial space is probably a function of concentration gradients, as well as endothelial permeability.9,73 Normal intima does not retain LDL particles, thus suggesting that most particles return to plasma or are degraded in situ. However, subendothelial retention of apoB-100–containing lipoproteins is an early event in atherosclerosis.73 Factors favoring net retention include a balance of uptake and degradation of LDL by macrophages, egress of LDL-containing macrophages into the circulation, and complex interaction with proteoglycans such that the LDL particles are sufficiently modified to maintain the gradient. Once bound to the matrix, the lipoprotein is modified so that it becomes oxidized.9,74 Oxidized LDL, unlike native LDL, is chemotactic to monocytes and rapidly taken up by macrophages to form foam cells by scavenger receptors.
Monocytes
As early as 1958, Poole and Florey noted that macrophages adhere to the surface of endothelial cells overlying atheroma.46 In studies of hypercholesterolemic monkeys, monocytes are seen attached to the endothelium within 12 days of initiation of an atherogenic diet.19 It is now apparent that monocytes play a very early role in atheroma formation.75,76 Endothelium, which is rendered activated, expresses adhesion molecules that interact with circulating leukocytes, principally monocytes and lymphocytes. E- and P-selectins slow monocytes, mediate rolling, and loosely tether them to the endothelium.77 More permanent fixation is due to members of the immunoglobulin superfamily—vascular cell adhesion molecule-1 (VCAM-1)78 and intercellular adhesion molecule-1 (ICAM-1)—which are soon upregulated and firmly fix the leukocytes to the wall. The importance of the selectins, VCAM-1, and ICAM-1 in the pathogenesis of atherosclerosis is evident in either ICAM-1 or VCAM-1 mutant animals exhibiting far less atherosclerosis than do wild type controls.79–81 The adherent leukocytes are stimulated to migrate into the subendothelial space by a number of chemokines, including MCP-1 and oxidized LDL.82–84
Macrophages
Once within the subendothelial space, monocytes undergo a series of phenotypic modulations and become resident tissue macrophages that take up oxidized LDL via the scavenger receptor A (SR-A), as well as CD36.85 One of the key signals for macrophage activation, macrophage colony-stimulating factor, can enhance scavenger receptor expression and promote replication of macrophages and their production of proinflammatory cytokines. Experiments in mutant mice deficient in macrophage colony-stimulating factor have shown the importance of this factor in atheroma formation.86,87 Although most foam cells are macrophage in origin, SMCs also take up lipids via scavenger receptors and CD36.
In mice, proinflammatory subsets of macrophages called M1-macrophages, induced by hyperlipidemia, produce the inflammatory cytokines IL-1β and TNF-α. The IL-1 gene family encodes three major proteins. The first two, IL-1α and IL-1β, exert proinflammatory effects by binding to the IL-1 receptor type 1. The third is IL-1 receptor antagonist (IL-1Ra), an endogenous inhibitor that competitively blocks the binding of IL-1α and IL-1β to the IL-1 receptor. Direct evidence for the proatherogenic role of IL-1 was obtained in experiments in which apoE−/− mice received subcutaneous injection of recombinant IL-1 receptor antagonist. Mice injected with IL-1 receptor antagonist displayed a marked reduction in atherosclerotic lesion size. Similarly IL-18, previously called interferon-γ (IFN-γ)–inducing factor, is a Th1-promoting cytokine and therefore has the capacity to promote inflammation through the innate and the adaptive immune pathways.88,89 Knockout of IL-18 in atherosclerosis-prone animals reduces aortic atherosclerotic lesion size.
Macrophages also contribute to thrombosis in several pivotal ways. As discussed later, atherosclerotic lesions that have ruptured characteristically contain abundant macrophages underlying a thin and collagen-poor fibrous cap. Specific collagenases MMP-1, MMP-8, and MMP-13, co-localizes with macrophages in human atheroma. Rupture of the fibrous cap may be a balance between collagen synthesis by SMCs and collagen breakdown by interstitial (matrix metalloproteinases) collagenases generated by activated macrophages. Studies in collagenase-resistant mutant “knockin” mice crossbred with apoE−/− mice demonstrated increased intimal collagen and SMC content.90 Similarly, in compound mutant apolipoprotein E–null mice crossed with MMP-13/collagenase-3−/− mice have increased fibrillar collagen that is thicker and more aligned in aortic atherosclerosis.91 Hence interstitial collagenases produced by activated macrophages greatly influence the structure and integrity of the fibrous cap overlying atherosclerotic plaques. Macrophages and SMCs within atherosclerotic plaques also overexpress the potent procoagulant tissue factor in response to C-reactive protein or CD40 ligand. Hence inflammation links atherosclerosis and thrombosis, leading some to refer to it as atherothrombosis.
Lymphocytes and Adaptive Immunity
The finding in the early 1980s that macrophages expressed major histocompatibility class II antigens needed for antigen presentation to CD4+ T cells suggested that adaptive immunity was involved in the atherosclerotic process.92 T lymphocytes, which account for as much as 10% to 20% of the leukocyte population, are found most abundantly in the shoulder and fibrous cap region of the atheroma. In advanced lesions, these T cells display markers of chronic activation and produce the prototypical Th1 cytokine, (IFN-γ) which further stimulates expression of class II major histocompatibility antigens in SMCs and macrophages. The crossbreeding of severe combined immunodeficiency (scid)/scid mice that lack T and B cells with apoE−/− mice produces offspring that are both hypercholesterolemic and immunodeficient. Similar to what was found with apoE−/−/Rag2−/− double mutants, when lesions in these mice were compared with those in the immunocompetent apoE−/− animals, a dramatic 70% reduction of lesion size was observed. However, transfer of oxidized LDL-reactive T cells to apoE−/−/scid/scid mice is more efficient at lesion acceleration than the transfer of T cells with no specificity to a plaque-derived antigen, demonstrating the importance of antigen presentation in the pathogenesis of atherosclerosis.93
Because of the participation of all components of the innate and adaptive immune system, atherosclerosis resembles other inflammatory and autoimmune diseases such as rheumatoid arthritis and type 1 diabetes mellitus. Potential endogenous autoantigens to activate T cells include LDL or heat shock protein 60.93,94 Cellular and humeral immune responses are mounted toward these antigens in humans and mice, and protective immunization strategies in mice have provided encouraging results.
In mice, functionally distinct T cell subsets appear to exist in atheroma. Natural killer T cells appear to accelerate atherosclerosis when recognizing lipid antigens presented through CD1 molecules.95 Conversely, regulatory T cells may suppress Th1 effector cells through production of antiinflammatory cytokines such as IL-10 and transforming growth factor-β (TGF-β).96 T cells, although far fewer in number than macrophages, likely serve as key regulators of the concerted immune response.
Mast cells are located at the shoulder and more central area of the cap, where they may participate in rupture or erosion of the cap.97 Once recruited to the subendothelial space in the intima, the white blood cells can perpetuate and amplify the ongoing inflammatory response that led to their recruitment.98 The CD40 receptor and CD40 ligand are expressed by several inflammatory cells, including macrophages and B and T lymphocytes.99 It is thought that this system contributes to leukocyte adhesion, matrix degradation, and cytokine-induced inflammation. Interruption of the CD40 signaling pathway reduces progression of atherosclerosis in experimental models.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


