(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
In this introductory chapter to Mitochondria Role in Cardiovascular Diseases, we will discuss those primary defects in mitochondrial and nuclear genomes that cause alterations in major aspects of mitochondrial metabolism. They include defects in OXPHOS and TCA cycle activity and regulation, mitochondrial membrane proteins, channels and transporters, transcription, translation and posttranslation modification factors, mitochondrial ribosomal proteins, mtDNA replication and repair, as well as mitochondrial dynamic. How these defects contribute to pathogenesis of cardiovascular diseases will be described in detail later in dedicated chapters.
The Energy-Consuming Heart
The human heart contracts at a rate of approximately 72 beats per minute 100,000 times per day pumping almost 10 tons of blood to ensure oxygenation of all tissues of the body. To maintain permanent rhythmic contractions, the heart requires constant generation of energy. Hence, it is not surprising that the myocardium is one of the most energy-consuming tissues. The energy, which is necessary to perform heart function, is derived from hydrolysis of the high-energy compound, adenosine triphosphate (ATP). The human heart consumes approximately 6 kg of ATP every day. Inability to generate a sufficient amount of cardiac energy plays a main role in heart failure (HF) [1–3]. HF is a complex multifactorial disease, which affects more than 2% of the US population, and is associated with an annual mortality rate of 10% [4, 5]. In the United States, treatment for HF costs approximately $28 billion annually [6]. ATP utilized by the myocardium is produced by mitochondrial metabolism, mainly by mitochondrial oxidative phosphorylation (OXPHOS). This process occurs on the inner membrane of mitochondria and couples electron transfer and oxygen consumption with phosphorylation of ADP into ATP. Mitochondria occupy 20–30% of the cell volume of a cardiomyocyte highlighting their critical role in the high demand of energy in the heart. Moreover, their numbers can significantly increase in response to the cardiac energy needs through the complex process of mitochondrial biogenesis.
The Mitochondrion: Origin, Morphology, Composition, and Dynamics
The term “mitochondrion” originated from the Greeks words “mitos,” thread, and “chondros,” grain reflecting the typical shapes of mitochondria first identified by light microscopy more than 120 years ago [7]. The readers are referred to a number of excellent reviews and textbooks to follow the history of the discovery and evolution of mitochondria [7–10].
The presence of the nucleus, containing elaborately organized chromosome and molecular machineries responsible for replication and transcription of the chromosomal DNA, and mitochondria, mainly responsible for cellular energy metabolism, is the major distinguishing features of eukaryotic cells. According to the endosymbiotic theory, mitochondria evolved from Gram-negative bacterial progenitors 1.5–2 billion years ago [11–15]. The rise of oxygen in the Earth’s atmosphere, some 2.4 billion years ago, forced an ancestor of eukaryotic cells to acquire oxygen-related organelles, such as peroxisomes, mitochondria, and plastids [14, 16]. An α-proteobacterial ancestor invaded and resided inside a primitive eukaryotic-like “host” cell, as an endosymbiont, and was eventually converted into cellular organelles, mitochondria, and plastids. It is believed that continuous selective pressure on the host cell to optimize energy production by the endosymbionts resulted in the establishment of nuclear genes which controlled many functions of the captive prokaryotes [9, 17]. In the process, endosymbionts lost the bulk of their genomes, autonomy, and much of their eubacterial identity; acquired many host features; and evolved into contemporary organelles.
Advances in imaging techniques, the development of high resolution fluorescent microscopy and mitochondrial specific fluorescent probes, electron microscopy (EM), and electron tomography, have led to a revolution in mitochondrial research. It becomes evident that mitochondria in living cells can interchange their morphology between elongated interconnected mitochondrial networks and fragmented disconnected organelles. These highly dynamic rearrangements are generated by the processes of mitochondrial fusion and fission and are regulated by specific fusion and fission proteins, respectively [18–20]. Emerging evidence suggests that changes in mitochondrial morphology may be an important factor in the pathophysiology of myocardial ischemia/reperfusion injury and heart failure (HF).
Transmission EM has shown mitochondria as double-membrane enclosed organelles with an inner membrane forming numerous invaginations, cristae. In mammalian cardiomyocytes, mitochondria are localized in longitudinal rows within the contractile apparatus, so-called interfibrillar mitochondria (IFM), or in clusters beneath the sarcolemma, so-called subsarcolemmal mitochondria (SSM) [21]. These two subpopulations of cardiac mitochondria differ in the structure of their cristae: IFM have tubular cristae, whereas SSM have lamelliform cristae [22]. Cristae significantly increase the surface area of the inner membrane, and their number is markedly elevated in highly respiratory-active cardiomyocytes (i.e., threefold more than found in hepatocytes).
While most biological membranes have a roughly 50:50 ratio of protein to lipid, the inner mitochondrial membrane is characterized by a higher protein content exhibiting a protein to lipid ratio of 75:25. In cardiac tissue, the anionic phospholipid, cardiolipin, is particularly prevalent in the mitochondrial inner membrane, where it contributes to the function of various mitochondrial proteins. The voltage-dependent anion channel (VDAC), also known as porin, is the most abundant protein in the outer mitochondrial membrane. It facilitates the transport of ATP/ADP across the outer membrane [23, 24]. The mitochondrial permeability transition pore (MPTP), a non-specific channel permeable for most small molecules up to 1.5 kDa, mediates the increases in the permeability of mitochondria linked to cell death contributing to CVD [25–28]. However, the precise molecular structure of the MPTP remains to be determined. In the inner membrane, the components of the electron transport chain (ETC) and the F0F1 ATP synthase together with transport systems for adenine nucleotides and inorganic phosphate (Pi) are assembled.
The mitochondrial matrix compartment enclosed by the inner membrane contains a compact genome; circular, double-stranded mitochondrial DNA (mtDNA); RNAs; their own ribosomes; and a wide range of proteins. Mitochondrial proteins are implicated in a variety of essential processes, ranging from OXPHOS, the Krebs cycle, and fatty acid oxidation (FAO) to transport of substrates, ions, and nucleotides through mitochondrial membranes, signal transduction, and apoptosis.
The number and morphology of mitochondria in the cardiomyocytes can change in response to diverse physiological stimuli (e.g., exercise, hormones/cytokines, electrical stimulation) and pathophysiological insults (e.g., cardiac hypertrophy, HF). Under certain conditions, more than 1,000 mitochondria can be present in a cardiomyocyte. They are typically distributed along the entire length of the myofibrillar apparatus to provide a constant supply of ATP to the sarcomeres and have variable shapes likely related to the extent of mitochondrial fusion and fission (Fig. 1.1). The movement of mitochondria within the cardiomyocytes has been demonstrated; however, its mechanism is currently poorly understood.
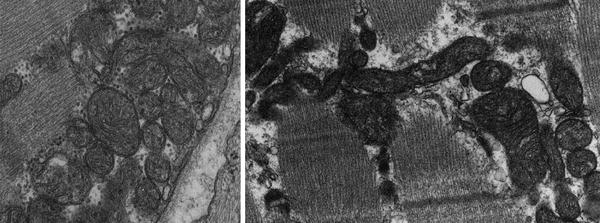
Fig. 1.1
Mitochondria of different shapes are distributed along myofibers
Cardiac Energy Metabolism
Cardiac energy metabolism is a complex process composed of three major pathways schematically shown in Fig. 1.2 [2]. The first pathway represents the breakdown of substrates, mainly free fatty acids (FA) and glucose (resulting from metabolizing food uptake), by β-oxidation and glycolysis. In the healthy heart, FA catabolism provides almost 90% of the ATP production, while glucose oxidation accounts for 10–40% [29]. The contribution of glucose to energy production is elevated by insulin during exercise [30]. Both β-oxidation and glycolysis lead to generation of an intermediate metabolite acetyl coenzyme A (CoA) that is utilized in the Krebs cycle, also known as the tricarboxylic acid (TCA) cycle. Enzymes implicated in the TCA cycle reside in the mitochondrial matrix and are linked to the inner membrane via succinate dehydrogenase (Complex II). The Krebs cycle results in the production of CO2 and NADH (nicotine adenine dinucleotide, reduced), the latter is used in the second pathway—mitochondrial OXPHOS. In this pathway, respiratory chain complexes I–IV, embedded in the inner membrane cristae, transfer electrons from NADH to O2 generating a proton electrochemical gradient (Δμ H+) across the inner mitochondrial membrane (protons outside and hydroxyl ions inside). The formed gradient drives the synthesis of ATP from ADP and Pi catalyzed by the F0F1 ATP synthase (complex V). Finally, the third pathway, the creatine kinase energy shuttle, transfers energy in the form of ATP to its consumption by myofibrillar ATPase and other ATP-consuming processes [31–33]. Creatine is not generated in the heart; it is produced in the liver and kidneys and transported into the cardiomyocytes through a specific plasma membrane creatine transporter [34]. Thus, cardiac energy metabolism converts chemical energy stored in our food into the mechanical energy of heart muscle contractions.
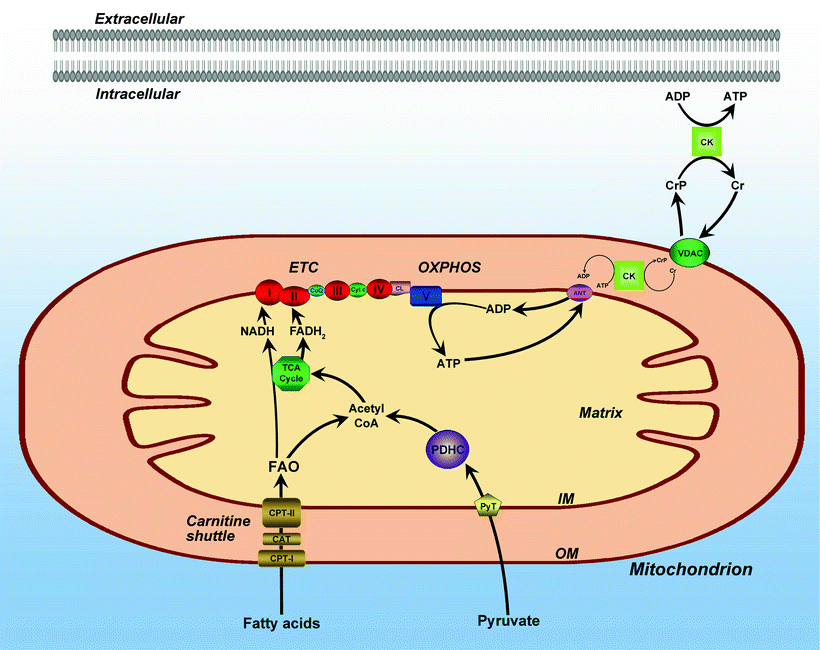
Fig. 1.2
Mitochondrial bioenergetic pathways. Intersection of mitochondrial bioenergetic pathways including fatty acid β-oxidation, pyruvate oxidation, oxidative phosphorylation (OXPHOS), and tricarboxylic acid (TCA) cycle. The electron transport chain (ETC) is composed of inner membrane (IM) localized respiratory complexes I–V with associated electron transfer components, coenzyme Q (CoQ), and cytochrome c (Cyt c) as shown. Also shown are the matrix associated pyruvate oxidation by pyruvate dehydrogenase complex (PDHC), fatty acid β-oxidation (FAO), and TCA cycle pathways. Also depicted: the carnitine shuttle pathway for the mitochondrial import of fatty acids, including carnitine palmitoyltransferases (CPT) I and II and carnitine-acylcarnitine translocase (CAT); the pyruvate transporter (PyT); and mitochondrial ATP/ADP transporting system, including adenine nucleotide transporter (ANT), voltage-dependent anion channel (VDAC), and creatine kinase (CK)-based energy transporting phosphocreatine shuttle. Other abbreviations: CL mitochondrial IM phospholipid cardiolipin; CoA coenzyme A; Cr creatine; CrP creatine phosphate; OM mitochondrial outer membrane
The heart demands for ATP production are tightly modulated not only during physiological transitions and perturbations, but also during cardiac development. Variations in mitochondrial morphology, substrate utilization, enzyme, and membrane activities serve to support developmental changes in cardiac function [35]. Since a comprehensive discussion on mitochondrial changes in the aging heart is presented in Chapter 13, here it is suffice to say that emerging evidence suggests an essential role for cardiac mitochondria in the complex process of the aging heart [36–39]. Mitochondrial OXPHOS is significantly decreased in the aging heart impairing myocardial metabolism [40]. While aging downregulates cardiac mitochondrial energy production, it increases the generation of reactive oxygen species (ROS), which are toxic to the cells. Among the multiple metabolic changes that occur in cardiomyocytes with advancing age are modifications in membrane composition, including increased levels of saturated FA and decreased levels of polyunsaturated FA and cardiolipin [41, 42]. Cardiolipin is a phospholipid unique to ATP-producing membranes, which is present in the healthy heart exclusively in the mitochondria, predominantly in the inner membrane [43, 44]. It plays a central role in OXPHOS supercomplex stabilization, mitochondrial membrane transport, ceramide biosynthesis, apoptosis, and aging [45, 46]. In addition, marked reductions in carnitine and acetyl carnitine levels have also been reported in the aging heart.
Mitochondrial Biogenesis
Mitochondria biogenesis is a complex, tightly regulated process, which is controlled by the mitochondrial and nuclear genome. The mitochondrial genome is packaged into DNA-protein particles termed nucleoids. Each nucleoid, a heritable unit of mtDNA, usually contains several copies (up to 10 in human cells) of mtDNA and various nucleoid-associated proteins [47–49]. Although the function of most nucleoid proteins has not been characterized, the high-mobility group (HMG) box family of nucleoid proteins plays an essential role in mtDNA packaging and maintenance. Mitochondria are able to replicate, transcribe, and repair their DNA.
Mitochondrial biogenesis requires the coordinated synthesis, import, and incorporation of various lipids and proteins. Growing evidence suggests that it is finely tuned largely at the level of transcription [50, 51]. The coordination between expression of mitochondrial genes in the nucleus and genes in mitochondria is mediated by an intricate network of transcription factors. Nuclear encoded transcription factors TFAM, TFB1M and TFB2M, RNA polymerase, and a termination factor mTERF control replication, transcription, and maintenance of mtDNA [52]. The nuclear respiratory factors NRF1 and NRF2 regulate expression of several nuclear-encoded mitochondrial proteins, including many components of the respiratory complexes I–V, while the peroxisome-proliferator-activated receptors (PPARs) govern expression of FA transporter and FAO enzymes [53, 54]. The estrogen-related receptors (ERRs) also contribute to the transcriptional regulation of uptake of energy substrates and ATP production and transport across the mitochondrial membranes [55]. The central upstream regulator of mitochondrial biogenesis and energy metabolism is the PPAR γ coactivator-1α (PGC-1α), which induces NRF1 and NRF2 transcriptional activities on the TFAM promoter, synchronizing thereby expression of mitochondrial and nuclear genome [56, 57].
Dysfunctional mitochondria are removed by autophagic destruction, mitophagy. Finely tuned balance between biogenesis and mitophagy is critical for mitochondrial quality control [58–60]. Genetic and environmental factors, alterations in metabolic signaling and oxidative stress, aging, and hyperglycemia can lead to reduced myocardial mitochondrial biogenesis and mitochondrial dysfunction contributing to cardiac disease.
Mitochondria, as the organelles responsible for generating ATP, have long been known as the cellular “powerhouse.” However, it is now increasingly appreciated that these complex organelles are involved in cardiomyocytes in a variety of additional processes, including generation of ROS and oxidative stress response, cell death, various signal transduction pathways, and intracellular Ca2+ homeostasis (Fig. 1.3) [61, 62].
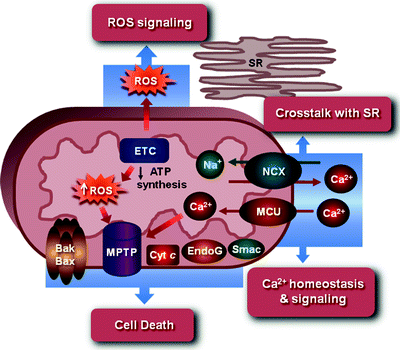
Fig. 1.3
Mitochondria as an integrator of ATP production, ROS generation and signaling, cell death, and Ca2+ homeostasis and signaling. See text for the details. Bak and Bax, Bcl-2 family proapoptotic proteins; Cyt c cytochrome c; EndoG endonuclease-G; ETC electron transport chain; MCU mitochondrial Ca2+ uniporter channel; MPTP mitochondrial permeability transition pore; NCX Na+/Ca2+ exchanger; ROS reactive oxygen species; Smac Smac/DIABLO proapoptotic protein; SR sarcoplasmic reticulum
ROS are produced in multiple cellular compartments by various pathways; however, approximately 90% of cellular ROS are generated as the by-products of mitochondrial OXPHOS. Unpaired electrons derived from NADH or FADH during mitochondrial respiration can react with molecular O2 to generate O2 •− [63–65]. Formed O2 •− can be converted into other ROS, such as hydrogen peroxide (H2O2), hydroxyl radicals (OH•), and ferryl radicals (Fe(VI) = O•− ) [66, 67]. These toxic cell-damaging oxidants are neutralized by the elaborate antioxidant defense system. Mitochondria possess several scavenger proteins, including manganese superoxide dismutase (MnSOD, also known as SOD2), glutathione (GSH), thioredoxin, peroxidase, and catalase [65, 68, 69]. The counterpart to the mitochondrial MnSOD is Cu/ZnSOD (SOD1) predominantly localized in the cytosol.
While the cell-damaging effects of ROS are well known, ROS are normal by-products of mitochondrial OXPHOS, and their physiological levels are important for normal cardiomyocyte function. Mitochondria-generated ROS function as essential signals, which induce an array of intracellular cascades mediating cell death, cardioprotection, cellular development, and proliferation [69].
Under physiological conditions, mitochondria maintain a tight balance between generation and scavenging of ROS to keep their levels within a physiological range. However, under pathological conditions, generation of ROS exceeds the capacity of the antioxidant defense system leading to detrimental effects on cardiomyocytes. Increased ROS levels can damage mitochondria resulting in further increase in ROS generation and leading to a vicious cycle of impaired mitochondrial function and the exacerbated ROS-induced cardiomyocyte damage [70, 71]. Mitochondria-generated ROS cause oxidative damage in the inner membrane phospholipids and proteins including subunits of ETC complexes. Oxidative alterations of ETC complexes lead to decrease in OXPHOS and eventually to the degradation of the damaged complexes as it has been observed in severe HF. ROS targets also mitochondrial and nuclear genome causing various DNA defects. mtDNA is particularly vulnerable due to its close proximity to the mitochondrial respiratory chain, the main producer of ROS.
Cardiac Mitochondria and Cell Death
Cardiac mitochondria play a pivotal role in three major pathways of cell death: apoptosis, necrosis, and autophagy [72–77]. All these cell death pathways occur in cardiomyocytes contributing to ischemia/reperfusion injury, myocardial infarction, and HF. Role of mitochondria in apoptosis associated with cardiac disease is the most characterized. Apoptosis is mediated by two major pathways; both pathways, while initiated by different stimuli, converge on the activation of cysteinyl aspartate proteases, caspases, which degrade myriads of cellular proteins ultimately leading to the death of the doomed cardiomyocyte (Fig. 1.4).
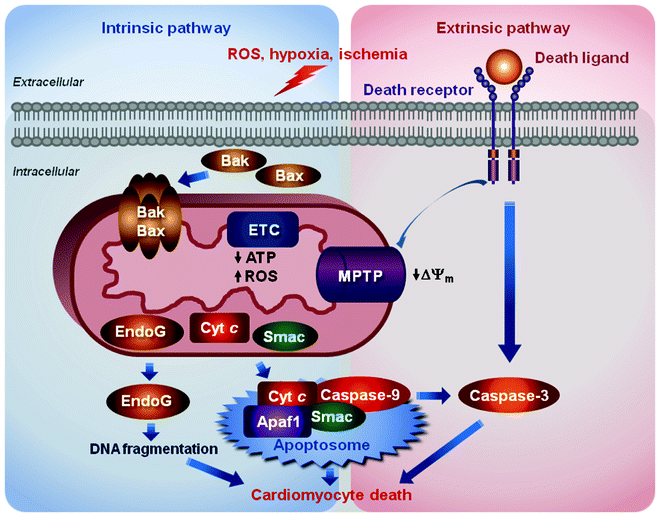
Fig. 1.4
Schematic representation of two major apoptotic pathways contributing to cardiomyocytes death. The intrinsic pathway is induced by diverse stimuli, e.g., oxidative stress, ischemia/reperfusion, and hypoxia, which activate translocation of the Bcl-2 family proapoptotic proteins, Bak and Bax, into the mitochondrial outer membrane. Bcl-2 family proteins permeabilize the outer membrane triggering the release of proapoptotic factors, cytochrome c (Cyt C), Smac/DIABLO (Smac), and endonuclease-G (EndoG), from the mitochondrial intermembrane space into the cytosol. Cyt C in association with the apoptotic protease-activating factor 1 (Apaf1) forms the multiprotein complex, apoptosome, in which procaspase-9 is activated to cleave and activate downstream caspases. Smac activates caspases through sequestering caspase inhibitors, while EndoG catalyzes the degradation of DNA. The extrinsic pathway is initiated by binding death ligands to their receptors resulting ultimately in activation of caspase-3, the key “executioner” caspase. These events trigger also the opening of mitochondrial permeability transition pore (MPTP) resulting in the dissipation of the mitochondrial membrane potential (ΔΨm), inhibition ATP synthesis, increased reactive oxygen species (ROS) generation, and eventually in swelling and rupture of the mitochondrion
The intrinsic pathway, also called the mitochondrial or Bcl2-regulated pathway, is induced by diverse stimuli, e.g., oxidative stress, ischemia/reperfusion, and hypoxia, which activate translocation of the proapoptotic members of the Bcl2 family, Bak and Bax proteins, into the mitochondrial outer membrane. Bcl2 proteins by not fully understood mechanism permeabilize the outer membrane triggering the release of proapoptotic factors, such as cytochrome c (the only soluble component of ETC), Smac/DIABLO, and endonuclease-G (endoG), from the mitochondrial intermembrane space into the cytosol. Cytochrome c in association with the cytosolic apoptotic protease-activating factor 1 (Apaf1) forms the multiprotein complex, apoptosome, in which procaspase-9 is activated to cleave and activate downstream caspases. Smac/DIABLO activates caspases through sequestering caspase inhibitors, while endoG catalyzes the degradation of DNA.
The extrinsic or death-receptor apoptotic pathway is initiated by binding death ligands to their receptors resulting in the death-inducing signaling complex (DISC)-mediated activation of caspase-8. Caspase-8 in turn ultimately cleaves and activates caspase-3, the key “executioner” caspase, as well as Bid protein. The latter triggers the opening of MPTP resulting in the dissipation of the ΔΨm, inhibition ATP synthesis, increased ROS generation, and eventually in swelling and rupture of the mitochondrion. Mitochondrial damage leads to the release of the above-described proapoptotic proteins. Cross-talk between these two apoptotic pathways is mediated by the truncated form of BH3-only protein Bid (tBid), cleaved by caspase-8. Under conditions, when the stress is prolonged and severe, ATP needed for apoptosis can be depleted significantly and necrosis will be induced [26, 27, 78, 79]. In addition to MPTP, mitochondrial ion channels, such as VDAC and K+ channels, adenine nucleotide translocase (ANT), cyclophilin-D, and connexin-43, are implicated as essential regulators of cardiomyocyte death.
Mitochondria and Ca2+ Signaling: Link Between Myocardial Contraction and Cell Death
Ca2+ is intimately involved in regulation of a number of vital cellular functions, including ATP production, gene transcription, muscle contraction, and cell proliferation and death. Ca2+ signaling plays a key role in cardiomyocyte physiology, and its deregulation is one of the central mechanisms underlying cardiac disease. The cardiac action potential triggers Ca2+-induced Ca2+ release from the sarcoplasmic reticulum (SR) to activate contraction elements [80–83]. Steady-state Ca2+ flux balance is tightly regulated and maintains by pumping back the released Ca2+ into the SR mediated by SR Ca2+-ATPase (SERCA2a) and Na+/Ca2+ exchanger. Mitochondrial Ca2+-permeable channel, known as the mitochondrial Ca2+ uniporter (MCU), also participates in uptake of released Ca2+. Moreover, mitochondria contribute to intracellular Ca2+ buffering by providing ATP, which is required not only for cardiac contraction but also for SERCA2a function [84–86]. Spatial organization of mitochondria within cardiomyocytes facilitates their interaction with the SR underlying a mechanism for mitochondrial and SR Ca2+ cycling cross talk [87, 88].
In mitochondria, Ca2+ can target multiple processes, including the TCA cycle, OXPHOS, and MPTP opening. Changes in mitochondrial Ca2+ levels modulate activity of F0F1 ATPase, ANT, Ca2+-sensitive dehydrogenases involved in the TCA cycle, and cytochrome c oxidase [89–94]. Mitochondrial Ca2+ overload mediates inhibition of major mitochondrial scavengers resulting in elevated ROS levels [94, 95]. Ca2+ overloading combined with increased ROS generation induces MPTP opening leading to the dissipation of the ΔΨm, uncoupling of OXPHOS, swelling and rupture of mitochondria, and releasing of death factors ultimately triggering cardiomyocyte death by apoptosis or necrosis. These processes have been involved in the pathogenesis of cardiac ischemia/reperfusion, cardiomyopathy, and HF [74, 76, 77].
Mitochondrial Dysfunction Related to Cardiovascular Disorders
Given the importance of mitochondria as the integrators of cellular energy metabolism, signal transduction, and cell survival, it is not surprising that mitochondrial dysfunction is associated with various human disorders, ranging from neurodegenerative diseases to cardiovascular diseases and cancer. Multiple alterations of cardiac mitochondrial metabolism have been implicated in pathogenesis of ischemia/reperfusion, HF, dysrhythmias, and atherosclerosis.
The concept of “mitochondrial medicine” was introduced in 1962 by Luft et al., who demonstrated a correlation between abnormal mitochondrial OXPHOS and clinical features in a case of severe hypermetabolism of nonthyroid origin [96]. Progress in the understanding of mitochondrial metabolism in the next two decades led to a general classification of the mitochondrial diseases [97]. In 1988, the molecular era of mitochondrial medicine initiated, when the first mutations in mtDNA were identified to be associated with human disease [98, 99]. Mitochondrial diseases represent a highly heterogeneous group of disorders, since they are caused by defects in both mtDNA- and nuclear DNA-encoded genes. It is believed that mutations in mtDNA account for approximately one-third of pediatric-onset and two-third of adult-onset mitochondrial disorders [100]. While initially these diseases were considered rare, progress in our understanding of mitochondrial function has led to estimations that 1 in 5,000 children develops a mitochondrial disorder and 1 in 200 adults carries a mtDNA mutation [101, 102].
In the past two decades, it has been identified multiple primary defects in mitochondrial and nuclear genomes causing alterations in major aspects of mitochondrial metabolism. They include defects in OXPHOS and TCA cycle activity and regulation, mitochondrial membrane proteins, channels and transporters, transcription, translation and post-translation modification factors, mitochondrial ribosomal proteins, mtDNA replication and repair, as well as mitochondrial dynamics [97, 103–105]. How these defects contribute to pathogenesis of ischemia/reperfusion, HF, dysrhythmias, and atherosclerosis will be discussed in detail in later chapters.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


