Chapter 6
Intimal Hyperplasia
Zhihua Jiang, C. Keith Ozaki
Based on a chapter in the seventh edition by Mark G. Davies
Intimal hyperplasia occurs frequently after vascular interventions such as vein bypass grafts, endarterectomies, arteriovenous fistulas, prosthetic bypass grafts, balloon angioplasty, endovascular stents and stent grafts, and solid organ transplantation. Data now suggest that restenosis develops in 5.8% of patients undergoing carotid endarterectomy or receiving carotid stents, though these cases rarely require re-intervention.1 About 30% to 60% of lower extremity arterialized vein grafts fail or are failing within a year,2,3 and intervention is frequently necessary to maintain conduit patency. Finally, 60% of hemodialysis access fistulas fail to mature primarily,4 and obliterative processes near the anastomosis are associated with half of these failures.5,6 Similarly, the durability of prosthetic dialysis grafts remains poor owing to intimal hyperplasia at the outflow vein.6,7 Although molecularly this process begins immediately after the vascular injury that accompanies the procedure, detectable intimal lesions classically form within a few weeks to 2 years.8
Definitions
Intimal Hyperplasia
The blood vessel intima is normally composed of just the thin endothelial cell lining of the vasculature. Intimal hyperplasia reflects additional biomass (cellular and matrix) within this layer. Mammals appear to have evolved intimal hyperplasic processes so that they become survival benefits. For example, hyperplastic intimal growth underlies closure of the ductus arteriosus. Vascular healing and premenopausal occlusion of the uterine arteries are also, in essence, the process of intimal hyperplasia.
Historically, the development of intimal hyperplasia was initially considered a “process of connective tissue hyperplasia.” Later, the concept was revised to “fibroproliferative” intimal thickening.9 Studies in the 1980s identified medial smooth muscle cells as central effectors of this process. Following biochemical insult and/or mechanical trauma, medial smooth muscle cells are activated and undergo a phenotypic switch from a contractile/quiescent to a synthetic/proliferative state. Proteases, particularly matrix metalloproteinases (MMPs), are released by inflammatory infiltrating leukocytes to break the matrix network. Smooth muscle cells are liberated from matrix restriction, and they then migrate from the tunica media to the intimal region, where they proliferate and deposit matrix proteins. Although sharing the same lineage identity with those in the media, smooth muscle cells in the intimal tissue are functionally incapable of organizing the cellular and matrix components. The de novo intimal layer is thus often qualitatively a relatively disordered histologic structure. This grossly fibrous, white material accumulates over time, protruding into the lumen and leading to luminal narrowing and even complete occlusion (Fig. 6-1).
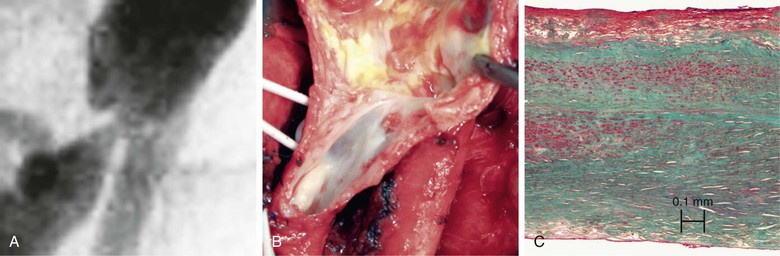
Figure 6-1 A, Digital subtraction angiogram of the distal right limb of an aortofemoral graft showing a filling defect at the junction of the prosthetic graft material and the outflow of the profunda femoris artery. B, Intraoperative photograph of the same field. Note the grossly fibrous, white intimal hyperplasia that corresponds to the outflow lesion. C, Masson trichrome stain preparation of endarterectomized intimal hyperplasia. Green staining corresponds to collagen, whereas red localizes primarily to other matrix components and the cytoplasm of largely smooth muscle cells.
It is currently believed that intimal hyperplasia starts as an acute inflammatory response. Because of the incomplete functional recovery of the repopulated endothelial cells and the phenotypic switch of smooth muscle cells, the acute inflammation often does not completely resolve, leaving behind a chronic inflammation in the intimal tissue. Inflammatory cells, particularly macrophages and T cells, are found in established intimal lesions. In contrast, atherosclerosis is a vascular disease driven by inflammatory mechanisms.10 The inflamed intimal tissue thus can serve as the “soil” for subsequent atherosclerosis to develop.11 Although the early failure of vascular reconstructions is largely caused by intimal hyperplasia, atherosclerosis is the dominant process underlying late failure (>3 years). After coronary artery stenting, atherosclerotic lesion(s) account for more than 70% of cases of recurrence of ischemic symptoms.12
Other Pathologies Underlying Lumen Loss after Vascular Procedures
Wall Remodeling
In addition to the intimal thickening, blood vessels can permanently change the dimensions of the media and adventitia. This change is termed wall remodeling.13 An early description of vascular remodeling comes from Glagov et al14 in 1987. They observed that coronary arteries actively grow to enlarge their cross-sectional area to accommodate the encroachment of atherosclerotic lesions. As the result of this compensatory expansion, appropriate luminal dimensions may be effectively maintained for the diseased vessels. This observation changed the concept of the vessels from inanimate pipes to an active system. It is now well established that the remodeling process involves reorganization/construction of both the medial and adventitial layers. Although leading to an increase in the overall dimension that favors preserving luminal area, remodeling may also proceed in the opposite direction, promoting wall shrinkage and loss of the luminal area.
Several terms have been used to describe remodeling processes. Morphologic terms, such as inward/constrictive remodeling and outward/expansive remodeling, are used to describe the impact on the geometric vascular dimension. Functional terms, such as positive remodeling and negative remodeling, were introduced to emphasize the effect of the remodeling process on the performance of the vessel as a conduit. A challenge in clinical studies is the inability to evaluate the full geometric dimension of the vessel over time because of the inadequate discrimination of the outer vessel wall from the perivascular tissue with imaging approaches such as angiography and computed tomography (CT) scans. This issue has been partially overcome by the availability of the intravascular ultrasound. Wall detail may be described in vivo as long as it is accessible by intravenous ultrasound probes.
Hemodynamic forces, particularly shear stress and wall tension, appear to be primary modulators of the vascular remodeling process. An elevated shear, for example, generally propels an expansion or outward growth, whereas a reduction in shear promotes “shrinkage” or inward growth of the vessel.15 Compared with that of shear forces, the impact of tensile forces on vascular remodeling is less well understood. Generally, wall tension is a factor that favors a negative remodeling process.16 When coupled with intimal hyperplasia, wall remodeling can ameliorate or exacerbate luminal narrowing through its effect on vessel dimension.
Vascular Tone and Recoil
Muscular vessels (arteries and veins) receive autonomic innervation from the central nervous system. Under physiologic conditions, a basal level of activity is transmitted via the sympathetic fibers to maintain a partial state of contraction in blood vessels. This basal level of contractile tension is referred to as vascular tone. Local hemodynamic forces and activation of vascular innervation may finely tune the vascular tone by way of promoting smooth muscle cell contraction or relaxation. Although transient fluctuation in vascular tone is reversible, a persistent increase in vascular tone may lead to structural changes such as the pathology occurring in patients suffering from long-standing arterial hypertension.
In addition to the active adjustment of the vascular caliber by smooth muscle cells, passive contraction of the vascular wall matrix network, consisting of predominantly collagen and elastic fibers, is also a critical determinant of the vessel diameter. Under physiologic conditions, the elastic resilience works to smooth out the pulsatility generated by cardiac ejection. Following the systolic distention, vessels return to their original dimensions as the result of elastic contraction. This phenomenon is termed vascular recoil. In the vessel wall, the intact and well-organized structural components as well as the blood counterbalance the wall shrinkage caused by recoil. In cases in which the wall integrity is destroyed, such as balloon angioplasty, recoil may cause 50% of the loss in acute gain during angioplasty.17,18 To oppose this recoil effect, metal stents were first used in peripheral arteries in 1985,19 and coronary arteries in 1986.20 In comparison with balloon angioplasty alone, the additional placement of stents can offer significant geometric benefits, as reflected by more preservation of the acute gain in lumen diameter and, in selected patients, less frequent requirement for secondary interventions and better quality of life.21
Restenosis
Several months after interventions such as endarterectomy, angioplasty, and bypass, the vascular constructions may become significantly narrowed or may occlude. This recurrence of stenosis and reduction in luminal area is termed restenosis. The causes for restenosis are complex.22 Usually, the primary pathology detected in a vessel with restenosis is intimal hyperplasia, and the loss of luminal area is proportional to the encroachment of the intimal tissue. Geometric remodeling likely also contributes.23 For example, the lumen area loss may be partially or completely compensated if a simultaneous outward remodeling had led to an increase in the geometric dimension. Conversely, significant restenosis may occur because of additional lumen loss caused by inward remodeling even if the amount of intimal hyperplasia is relatively small. Metal stents offer a strategy to limit inward remodeling, although intimal hyperplasia (either through the stent structure or at the ends of a stent graft) often offset these luminal area gains. Under specific circumstances, an improved long-term patency of the stented vessels has been observed.21
Inciting/Modulating Factors for Intimal Hyperplasia
Intimal hyperplasia is a vascular response that heals the injured vessel wall. It can occur in nearly every type of vascular reconstruction, and the initiation and progression of the lesion may be modulated by several factors.
Hemodynamic Stress
Hemodynamic forces, specifically shear stress and wall tensile stress, are well-established initiators and modulators of intimal hyperplasia.24 Vascular cells are equipped to sense and respond to the luminal hemodynamic environment. Several surface receptors (e.g., integrins and vascular endothelial growth factor receptor [VEGFR]), ion channels (e.g., K+ and Ca++), and the cell cytoskeleton may be specialized in mechanosensing and mechanotransduction.25
Under physiologic conditions, the steady laminar blood flow generates on average approximately 15 dyne/cm2 of shear stress in arteries. Endothelial cells sense this physiologic shear force, releasing mediators such as nitric oxide (NO) and Kruppel-like factor-2 to maintain a quiescent state for smooth muscle cells and homeostasis of the whole vessel wall.26 Vascular reconstructions such as vein bypass grafts, stented diseased arteries, and arteriovenous fistulas not only alter the rate of the local blood flow but also frequently induce a disordered flow pattern. Both clinical and experimental observations have demonstrated that disturbed flow and/or low wall shear stress accelerate development of intimal hyperplasia.15,27,28 Endothelial cells respond to these particular hemodynamic conditions by elaborating adhesion molecules, proinflammatory cytokines, and other bioactive substances that in turn enhance cell proliferation and matrix accumulation, leading to robust intimal growth.29,30 On the other hand, laminar, high blood flow (uniform high shear stress) generally exhibits an opposing effect on intimal growth. For example, vein grafts exposed to high flow conditions tend to develop less intima than those with low blood flow.15 Furthermore, augmentation of blood flow in vessels with established intimal hyperplasia induces intimal regression.31 Although the exact mechanisms remain to be fully elucidated, experimental studies suggest that high laminar shear shifts the local vascular milieus from a proinflammatory to an antiinflammatory state, imposing a differential regulation of intimal growth.26 This knowledge has been translated to clinical application, so that creation of a distal fistula to boost the blood flow has led to improvement in the patency rate of lower extremity grafts in selected circumstances.32 Although generally protective, shear may be deleterious when it reaches an extremely high level or becomes disordered.33 Finally, compared with the well-established effect of shear stress, the impact of tensile force on intimal hyperplasia is much less defined. In the cell culture setting, mechanical stretch activates several pathways capable of regulating smooth muscle cell phenotype.34,35 Evidence in vivo suggests that tensile force correlates positively with intimal thickening.16,36
Immune Insults
Solid organ transplantation usually introduces a mismatch in the HLA complex between the transplant and the host immune system, triggering immunologic responses that accelerate formation of diffuse, concentric intimal hyperplastic lesions in the arteries of the allograft that may eventually result in allograft failure. Reflective of this complex immune scenario, allograft vasculopathy is more vulnerable to in-stent restenosis after intervention than the stenotic lesions in native coronary arteries.37 Multiple cell groups, particularly T-cell subsets (CD4+ and CD8+) and B cells, appear to be critical to the pathogenesis of these intimal hyperplastic lesions in the transplant setting. Activated by antigen recognition through either direct (major histocompatibility complex I [MHC I]–dependent) or indirect (MHC II–dependent) pathways, these cells may cause endothelial cell dysfunction via cytolysis, cytokine stimulation (interferon-γ [IFN-γ], interleukin-12 [IL-12]), and specific antibody production.38 Beyond the transplant setting, similar immunologic insult may occur in stented vessels, where immune cells are activated via a response to foreign bodies.39 Finally, risk factors for accelerated intimal hyperplasia identified in clinical studies (hyperlipidemia, diabetes, and smoking) may act by way of augmenting chronic inflammation in the vessel wall.
Genetic Susceptibility
In addition to environmental factors, genetic status stands as a risk factor for intimal hyperplasia. Some of this knowledge was acquired via genome-wide association studies (GWASs). For example, single-nucleotide polymorphisms (SNPs) in the IL-10 gene are associated with higher rate of restenosis in coronary arteries treated with drug-eluting stents.40 Susceptible loci in chromosome 12 have also been documented.41 These genetic discoveries have highlighted the horizon for genetic manipulations as a therapy for inhibiting intimal growth.
Biologic Mechanisms
Intimal hyperplasia is a highly complex process that involves several tissues (perivascular,42 vessel wall, and blood), numerous cell lineages, and multiple molecular signaling networks. Although much has been learned in the past few decades, many mechanistic details remain to be fully elucidated. The current paradigm is founded on the postulate that intimal hyperplasia is a vascular response to injury, with the cellular and molecular events generally mirroring those occurring in the course of wound healing.8,43
Cellular Effectors
Compared with the medial layer, the neointimal lesion contains relatively more extracellular matrix, with a disordered cell-matrix organization that mimics tissue fibrosis immediately beneath the intact endothelial cell lining. However, unlike reactive fibrotic tissue, which is primarily populated with fibroblasts, vascular intimal lesions are dominated by cells that express smooth muscle cell markers, such as α-actin and smooth muscle myosin heavy chain (SM-MHC). Work in the 1980s suggested that these cells are largely medially derived.44 However, in addition to derivations from the tunica media, later studies have identified cells from nonmedial sites, such as the adventitia and perivascular tissue,45 stem cell/progenitor cell niches,46,47 and the circulating blood as suppliers of the intimal cell population.
Once recruited to the intimal lesion, these cells gain a phenotype similar to those derived from the medial smooth muscle cells, making them indistinguishable from each other.48,49 Although vascular wall cells are normally relatively quiescent, the repopulated intimal cells often display a proliferative and synthetic state. Substances that are not produced basally in the vasculature (e.g., cytokines, growth factors, and adhesion molecules) are induced in these neointimal cells.50 Acting through autocrine and paracrine mechanisms, these substances maintain an inflamed state for the intimal tissue, collectively propelling progressive growth of the intimal lesion.
Endothelial Cells
The luminal surface of blood vessels is covered by a monolayer of endothelial cells. Under physiologic conditions, these cells serve as a “physical barrier” that segregates the circulating blood cells and molecules from tissue matrix components, and also function as a “secretory organ,” releasing an array of mediators to maintain homeostasis in the wall and beyond. Lining the luminal surface, endothelial cells produce transmembrane adhesion molecules such as vascular endothelial cadherin (VE-cadherin) and neural cadherin (N-cadherin) for intercellular connections. Interacting with the intracellular cytoskeleton, these molecules form adhesive junctions (e.g., tight junctions, gap junctions, and adherens junctions) with neighboring cells, allowing selective exchange of molecules between blood and tissue.51 Anticoagulants (e.g., heparin, thrombomodulin, prostaglandin I2 [PGI2], and Kruppel-like factor-2) are synthesized and released by these cells to maintain a relatively anticoagulant and thus thrombus-free luminal surface.
In addition to these “direct” biologic effects, endothelial cells govern the homeostasis of the vessel wall via communication with the underlying smooth muscle cells. A well-defined tool for endothelial cells to carry out this function is NO, which has been linked with intimal hyperplasia.52 NO is very lipophilic. Once released, it quickly diffuses into the medial layer and acts directly on smooth muscle cells. The half-life of the endogenous NO is so short (0.1-5 seconds) that the effect on smooth muscle cells can be rapidly turned on or off depending on demand. NO was initially identified as endothelium-derived relaxing factor because of its function to induce vasodilatation. Subsequent studies demonstrated that NO is also a key messenger for endothelial cells to keep the medial smooth muscle cells at a quiescent and contractile phenotype.53
Upon activation by forces such as altered hemodynamics or physical/chemical trauma, endothelial cells convert their transcriptome to a profile that is significantly different from that of their quiescent state.54 Leukocyte and platelet adhesion molecules are upregulated or translocate to the cell membrane. Genes encoding proinflammatory mediators (e.g., chemokines, cytokines, adhesion molecules) are induced or upregulated, whereas those encoding antiinflammatory mediators (e.g., NO, anticoagulants, Kruppel-like factor-2) are downregulated.55 These bioactive substances can trigger a series of events, such as smooth muscle cell activation, inflammatory cell infiltration, and the recruitment of vascular progenitor cells—all contributing to intimal hyperplasia. Finally, studies have now suggested a new mechanism whereby endothelial cells may contribute to intimal hyperplasia through endothelial-mesenchymal transition (EMT).56,57
Smooth Muscle Cells
The intimal hyperplasia tissue, just like the tunica media (though morphologically distinct), is dominated primarily by smooth muscle cells. During the development of intimal hyperplasia, intimal smooth muscle cells display a spectrum of phenotypes with preferential losses and gains of their differentiation markers at various stages. The initial dedifferentiation process leads to a loss of expression of the majority of the differentiation markers, such as α-actin, desmin, and smooth muscle isoforms SM1 and SM2. As the lesion becomes more established, early differentiation markers such as α-actin and desmin are gained relatively quickly. The reproduction of the more mature markers, such as smooth muscle isoforms SM1 and SM2, on the other hand, may be very much delayed.58,59
Although they normally function in the media primarily as the drivers for vascular relaxation and constriction, smooth muscle cells in the intimal lesions serve as effectors that produce cellular and extracellular components to expand the intimal mass. Having switched to the proliferative/synthetic state, smooth muscle cells respond to local cytokines and growth factors by vigorous cell division and matrix production. Experimental studies have demonstrated that the proliferation may start a few hours after an inciting event, reach a peak in a few days, and last for weeks to months.15,60 Genes encoding matrix components (particularly interstitial matrix proteins such as hyaluronic acid, chondroitin, collagens, proteoglycans, fibronectin, and elastin) are greatly upregulated. On the other hand, the production and the activity of the matrix degradation system, which is consisted of proteases (e.g., MMPs, tissue plasminogen [t-PA], urinary plasminogen activator [u-PA], and plasminogen) and their counterpart inhibitors (e.g., tissue inhibitors of metalloproteinases [TIMPs] and plasminogen activator inhibitor-1 [PAI-1]), is modulated to favor a selective deposition of matrix proteins,8,61 leading to an accumulation of de novo interstitial matrix with an altered composition.
In addition to their role of directly making intimal mass, smooth muscle cells synthesize and release a wide spectrum of mediators to the extracellular space.50 Cytokines (e.g., IL-1β and tumor necrosis factor-α [TNF-α]), chemokines (e.g., IL-8 and monocyte chemoattractant proteins [MCPs]), and growth factors (e.g., platelet-derived growth factor [PDGF] and transforming growth factor-β [TGF-β]) have been documented as intimal smooth muscle cell products. The exact profile of these mediators varies according to the stage of intimal hyperplasia development. These mediators may act on smooth muscle cells themselves and other cell lineages through autocrine and/or paracrine function. Although the early mediators serve to amplify the inflammatory response and favor smooth muscle cell migration and proliferation, driving a hyperplastic response with high cellularity,62,63 those released by smooth muscle cells in the established neointima facilitate a fibrotic process.60
Finally, vascular smooth muscle cells may lose their production of contractile proteins through dedifferentiation and acquire a phenotype that overlaps with another vascular cell group, the myofibroblasts. Studies of both systemic hypertension and pulmonary hypertension have demonstrated a critical role for the adventitia and adventitial fibroblasts in the structural remodeling and the resultant pathology of the vessel wall,64,65 and these cells may participate in intimal hyperplasia.66–68 Like medial and intimal smooth muscle cells, myofibroblasts likely stand as cellular effectors that produce and react to various bioactive substances, modulating structural remodeling and intimal growth in the vessel wall.69
Platelets and Leukocytes
Following vascular injury, platelets aggregate on activated endothelial cells and denuded areas to cover the collagens and other matrix components that are exposed to the blood. Fibrin is deposited, together with the activated platelets, forming an adhesive surface to capture circulating leukocytes from the blood stream.70–72 Inflammatory cells such as monocytes, eosinophils, and T cells home to the injured site. Although representing only a relatively small portion in the eventual intimal cell population, inflammatory cells have been identified as central modulators and potential key orchestrators of hyperplastic intimal thickening. For example, clinical studies have demonstrated a positive correlation between leukocyte count and activation with the severity of stenotic and restenotic coronary lesions.73,74 The level of inflammatory markers typically associated with leukocyte activity, particularly C-reactive protein, holds some correlation with clinical outcomes.75,76 Strategies blocking leukocyte adhesion or creating leukopenia significantly inhibit intimal thickening in various experimental settings.72,77 Through their robust ability to produce various inflammatory mediators, including chemokines, cytokines, and growth factors, leukocytes appear to amplify the acute inflammation, contributing to intimal thickening.78
Leukocytes consist of several subpopulations. The quantity of each subset in intimal lesions seems to be determined by the involvement of specific initiating factors and the stage of the pathology. For example, lesions that developed in the setting of hypercholesterolemia or after severe physical injury tend to hold more macrophages than those triggered solely by endothelial denudation.72 Otherwise, neutrophils dominate the inflammatory infiltrates after moderate injury such as endothelial denudation.79 There is also evidence that T cells stimulate intimal hyperplasia via promoting a helper T cell type 1 (Th1) response,80 but this mechanism may be more significant in the pathogenesis of allograft vasculopathy.81 Advances in immunology have led to appreciation of distinct roles for subsets of monocytes and macrophages in the vascular inflammatory response. “Patrolling” and “resident” monocytes have been identified in the peripheral blood.82,83 Upon extravasation into tissue, these cells follow varying pathways to differentiate into M1 and M2 macrophages, performing distinct roles in pathogenesis and tissue repair.84,85 For example, M2 polarization of the resident monocytes has been demonstrated to be indispensable to arterial remodeling triggered by hemodynamic stress.86 Recent research efforts have attempted to identify the key regulators for these processes so that interventional approaches may be developed to guide the kinetics of the inflammatory cell trafficking and intimal lesion progression.87
Progenitor Cells
In addition to smooth muscle cells, several other cell groups are recruited to neointimal lesions. Vascular progenitor cell lines released from bone marrow and other stem cell niches may home to intimal hyperplasia.46,49 Initial evidence suggests that smooth muscle progenitors are released from stem cell niches to the circulation.88 Not only can these cells gain smooth muscle cell markers but they also seemed at first to respond to exogenous stimulation much like smooth muscle cells from tunica media do.89 However, subsequent studies have demonstrated that cells from circulation and bone marrow lack the ability to produce a full repertoire of smooth muscle cell contractile proteins in the intimal lesion90 and that they tend to maintain a sustained inflammatory phenotype.91
A groundbreaking discovery has been the participation of endothelial progenitor cells in reendothelialization of the injured luminal surface.92 Experimental studies revealed that approaches facilitating the homing of these progenitor cells to the lesion site significantly accelerated the recovery of the endothelial cell monolayer and inhibited neointimal hyperplasia.93 Clinical observations support an association of the impaired endothelial progenitor cell function with higher rate of stenosis and restenosis.94 Because the endothelial progenitor cell is a relatively rare element in circulating blood, strategies such as capturing antibodies and adhesion molecules or growth factors to enrich endothelial progenitor cells at lesion sites are being evaluated.95,96
Biochemical Mediators
Studies of vascular wall gene expression have described induction and suppression of numerous genes over the course of the development and progression of intimal hyperplasia. A large number of molecular mediators have been implicated in experimental studies as promising therapeutic targets, and only a selected few are discussed here. Efforts to translate the genomic knowledge to patients have rarely improved the clinical outcome.2 Compelling evidence obtained from both experimental and clinical studies has demonstrated that intimal hyperplasia is a highly complex process.54 It is driven by many networks of molecules functioning with temporal and spatial variations over the course of lesion development, and most of these mediators have significant functional redundancy. Targeting a single molecule with therapeutic interventions has been demonstrated to be unlikely to alter the biologic consequence of such a complex system,2,3,30 although risk factors such as diabetes, smoking, and genetic variations are capable of modulating this system and accelerating intimal hyperplasia development. Further complexity is manifested through the specifics among intimal hyperplasia occurring in response to various vascular events (in-stent restenosis, vein bypass failure, postendarterectomy stenosis, etc.). Each likely holds unique considerations for preventive and therapeutic strategies.
Inflammatory Mediators
Injury to the vessel wall results in aggregation of platelets and activation of vascular cells (e.g., endothelium, smooth muscle cells, and fibroblasts). Once activated, these cells are robust manufactures of bioactive substances, triggering a complex mediator cascade that amplifies the vascular damage and the inflammatory response. P-selectin, which is normally stored in the a-granules of platelets and Weibel-Palade bodies of endothelial cells, is released.97 E-selectin is transcribed, translated, and transported to the surfaces of endothelial cells to facilitate leukocyte rolling as part of the homing process.98,99 Chemokines—monocyte chemoattractant protein-1, RANTES (regulated on activation, normal T cell–expressed and secreted), IL-8, interferon-γ–induced protein [IP-10], and stromal cell–derived factor-1 [SDF-1]—are mobilized to create a concentration gradient to guide leukocyte migration. Other adhesion molecules, such as intracellular adhesion molecules (ICAMs), vascular cell adhesion molecule (VCAM), and integrins, are released, mediating firm leukocyte adhesion. Inflammatory cytokines (e.g., tumor necrosis factor-α and IL-1β) that are normally absent in the vessel wall are produced by activated leukocytes, predominantly monocytes. These cytokines serve as a driving factor for intimal hyperplasia via their mitogenic functions on smooth muscle cells100 and proapoptotic properties for endothelial cells.100–102 In amplification of this traditional dogma, studies have now identified danger-signaling networks as a mechanism for cell activation and the initiation of the inflammatory response. Toll-like receptors (e.g., TLR- 2 and TLR-4) recognize the degraded tissue components, initiating the innate immune response.42 Strategies manipulating the activity of the danger signaling have succeeded in altering the eventual vascular morphology.103–105
In addition to the proinflammatory cytokines, antiinflammatory cytokines such as IL-10 and IL-19 are produced in lesion sites, and these cytokines appear to be protective against lesion progression. In vein grafts, for example, enhanced IL-10 expression is associated with less lesion formation.30,106 Pharmaceutical treatment with recombinant IL-10 has been found to inhibit intimal hyperplasia formed in injured arteries.107 Similar protective effects were also observed for IL-19.108 As part of the regulatory network in the inflammatory response, these counterbalancing entities are primarily produced by the same cell population as the proinflammatory cytokines, specifically macrophages. Through targeting STAT (signal transducer and activator of transcription) and nuclear factor-kappaB (NF-κB) signaling pathways, these molecules inhibit smooth muscle cell proliferation and further induction of other inflammatory mediators, suppressing the hyperplastic vascular response and, thus, intimal growth.107–109
Although this acute inflammation frequently comes to complete resolution in other tissues, it can transition to a chronic state in the vessel wall, perhaps driven by continued hemodynamic or foreign body insult. Studies of inflammatory events occurring in nonvascular tissues support that the stromal microenvironment defined by cytokines (e.g., IL-1β and IL-6) and chemokines (e.g., stromal cell–derived factor-1 and monocyte chemoattractant protein-1) dictate the inflammatory response of complete resolution or chronic inflammation.110 Lipid mediators such as the resolvins and maresins can also downregulate inflammatory systems.111–113 Conversely, in the vascular wall an inflammatory cascade model supports an IL-1β–based system whereby the inflammation is sustained via a self-stimulatory mechanism.114
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


