CHAPTER 11 Interstitial Lung Diseases
The interstitium includes the alveoli, the epithelial and capillary cells within the alveolar wall, the septal tissues, and the connective tissues that surround the vascular, bronchial, and lymphatic structures within the lung parenchyma. An inflammatory response may involve any or all of these structures with varying clinical and radiographic presentations. The pathologic response to the resulting immunologic insult permits a clinically useful, although not strict, categorization of ILD into those entities characterized by alveolitic and diffuse interstitial inflammation and those that result in a predominantly granulomatous pattern of disease (Box 11-1).
GRANULOMATOUS PATTERNS OF ILD
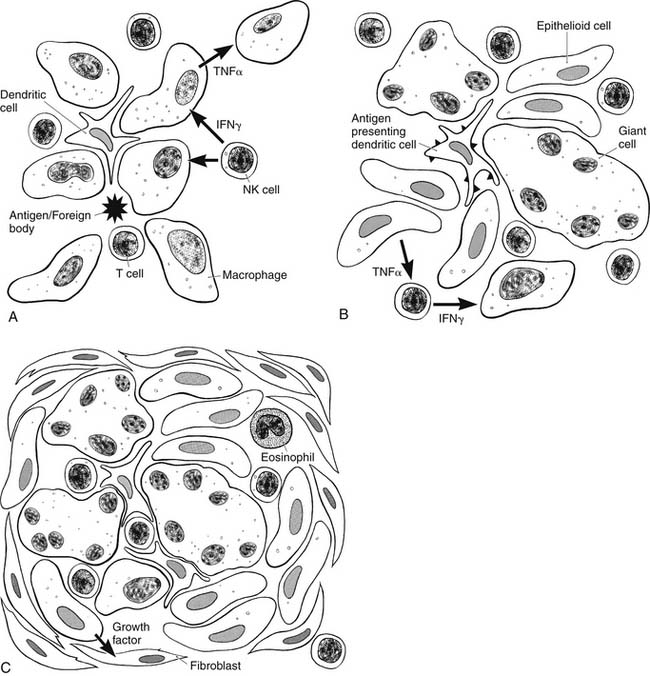
Granulomas consist of activated immune cells, typically macrophages, encircling particles that are generally not recognized by the antigen-specific, or adaptive, immune system. The particle can be a foreign body or an intrinsic protein antigen that activates the innate immune system. Innate immunity is a non–antigen specific means by which the body provides a first line of defense against foreign pathogens, including inhaled irritants. The innate immune system activates macrophages directly, without any need for prior exposure or antigen processing as is required for an adaptive T- and B-cell-dependent response. Activated macrophages mature to form epithelioid cells around the foreign material and thereby form a granuloma.1
However, macrophage activation and granuloma formation are also influenced by specific immune signals generated by other cells of the immune system, including signals triggered in an antigen-specific manner by the foreign particle or protein, as illustrated in Figure 11-1. Fragments of foreign proteins processed by antigen-presenting cells, such as dendritic cells, are displayed on the cell surface in the context of the host major histocompatibility complex (MHC). Through this indirect pathway, antigen-specific CD4+ helper T cells are activated in the context of class II MHC presentation, whereas CD8+ effector T cells traditionally require the class I MHC complex. These activated T cells produce various cytokines that trigger several antigen-specific and nonspecific cellular subsets. For example, the activated TH1 subsets of CD4+ helper T cells and natural killer (NK) cells produce interferon-gamma (IFN-γ), leading to primarily macrophage activation. On the other hand, the TH2 subset produces interleukin-4 (IL-4) and interleukin-5 and results in eosinophil production and activation.1–3 Other T-cell subsets, such as CD8+ effector cells, are also recruited to help clear the protein antigen and thus help regulate the immune system. Once activated, this inflammatory process results in the recruitment of other immune cells, complement, and growth factors to form discrete granulomas, or the process may spread to the alveolar spaces and walls, leading to alveolitic changes (described later). Tumor necrosis factor (TNF), a potent inflammatory cytokine, has been implicated in initiating and driving the inflammatory process in many of these pathologic states. Newer targeted therapies for many inflammatory ILDs involve anti-TNF therapies.4,5 Granulomatous mechanisms are involved in a variety of pulmonary diseases, yet they all share this fundamental pathophysiologic mechanism. The clinical presentations and findings of these disease entities are described later.
Foreign Bodies and Inorganic Dust
Patients who have inhaled foreign bodies are typically asymptomatic at the time of initial exposure unless the particle is large enough to occlude the tracheobronchial tree. In such cases, as often seen in children, the diagnosis is made by history, confirmed by chest radiography, and, if needed, diagnosed as well as treated via bronchoscopy. However, prolonged exposure to smaller foreign particles, whether organic or inorganic, can lead to a spectrum of pulmonary disease processes. Chronic exposure to metal dusts, such as beryllium, aluminum, and zirconium—foreign particles that cannot be cleared by the mucociliary system or broken down by the pulmonary macrophages—is associated with a marked interstitial granulomatous disease pattern.6 Continuous exposure to small organic particles in susceptible individuals can also lead to hypersensitivity pneumonitis, with both alveolitic and granulomatous responses. The effect of a one-time massive exposure to inorganic fine particulate matter is less clear. A large subset of the pedestrians and workers exposed to smoke, dust, and particulate matter after the World Trade Center attacks in 2001 presented with a variety of nonspecific respiratory ailments, often with documented nonspecific histologic changes on lung biopsy. However, their long-term outcomes are not yet known.7,8
Hypersensitivity Pneumonitis
Hypersensitivity pneumonitis, also known as extrinsic alveolitis, describes a pulmonary disease process characterized by immunologically induced injury of the lung parenchyma with granuloma formation, alveolitis, and an antibody response, as the result of repeated exposure to inhaled protein antigen.9–11 Numerous antigens have been implicated, from Aspergillus and fish meal dust to male rat urine, leading to various interesting, occupation-associated disease names (Table 11-1). Initial exposure to the antigen induces neutrophil and macrophage extravasation to the distal bronchioles and alveoli, thus leading to early alveolitis. Patients may present acutely with cough, fever, and chills several hours after exposure. With continued antigen exposure, patients develop a persistent cough with worsening dyspnea secondary to granuloma formation within the pulmonary parenchyma, and progressive interstitial disease.9,11
Table 11–1 Selected Hypersensitivity Pneumonitis Syndromes
| Syndrome | Antigen |
|---|---|
| Bagassosis | Actinomycetes species from sugar cane |
| Cheese washer’s lung | Penicillium casei from moldy cheese |
| Compost lung | Aspergillus species from compost |
| Furrier’s lung | Animal fur dust |
| Hot tub lung | Mold on ceilings |
| Laboratory worker’s lung | Rat urine |
| Fish meal worker’s lung | Fish meal dust |
| Woodworker’s lung | Wood dust |
The diagnosis lies in careful history taking, especially when a known occupational exposure is suspected. Chest radiographs have no diagnostic pattern, though may show a reticular nodular pattern consistent with interstitial lung disease. Chest computed tomography (CT) is typically nondiagnostic as well.10,11 Bronchoalveolar lavage (BAL) may be helpful, showing an increase in CD4+ T cells acutely and CD8+ T cells chronically. However, this narrows the spectrum only marginally, as this is characteristic of nearly all granulomatous disease processes. Serum tests for antibodies to the suspected antigen are often required to make a diagnosis. Lung biopsy, either transbronchially or via video-assisted thorascopic surgery (VATS) or open thoracotomy, may be necessary to make a diagnosis in those patients for whom other evidence is lacking. Even this aggressive approach, however, may be nondiagnostic in late stages when the acute granulomatous and alveolitic changes are replaced by interstitial fibrosis.
Treatment consists of removing exposure to the suspected antigen, and steroids for patients with the severe acute form of the disease or with chronic persistent fibrosis.10–13 Histologic evidence of fibrosis in lung biopsy specimens is correlated with a worse prognosis.13 Radiologic evidence of fibrosis seen on high-resolution CTs may serve as a surrogate for histologic findings once the diagnosis has been made, and this may correlate with long-term survival and response to therapy.12
Infections
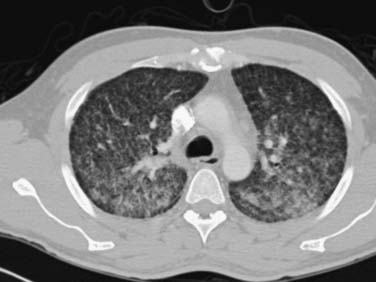
Pulmonary infection with Mycobacterium tuberculosis is probably the most common cause of granulomatous lung disease worldwide.2,14 Local as well as disseminated miliary disease leads to granuloma formation in the infected field (Fig. 11-2). Other infectious organisms such as Aspergillus and certain helminths can also lead to pulmonary granulomatous disease.2 Radiologic assessment alone is often inadequate to make a diagnosis, and the clinical context must be taken into account. The clinical pathogenesis, presentation, course, and treatment of pulmonary infections are discussed elsewhere in this book and are not included in this discussion of ILD.
Sarcoidosis
Sarcoidosis is a chronic systemic disorder that, although common, is still poorly understood. Various organ systems of these patients are often affected by granulomatous disease, and some individuals do not manifest pulmonary involvement.15–17 The incidence of sarcoidosis varies with the population studied. In Western populations, the incidence is estimated to be 10 to 20 per 100,000 population, with a higher rate in women and those of African-American ancestry.18 Sarcoidosis is uncommon in Asian populations. Although the disease can manifest at any age, most patients present between the ages of 20 and 40.15,16,19,20
The etiology of sarcoidosis is unknown. Environmental, infectious, hereditary, and immunologic factors have been postulated, with variable evidence to support each hypothesis.20 The interstitial granulomatous disease that results from inhalation of metal dusts is pathologically identical to sarcoidosis, suggesting that an unknown environmental exposure may be responsible.6,21,22 Supporters of an infectious etiology argue that the causative agent must be inhaled, because 80% of affected individuals have pulmonary and mediastinal lymph node involvement. In fact, BAL of individuals with extrapulmonary sarcoid contains inflammatory cells even without evidence of pulmonary disease. The causative agent is suspected by some to be Mycobacterium tuberculosis or a nontuberculosis mycobacteria. Genetic analyses of affected tissue by polymerase chain reaction as well as antibody analysis of afflicted individuals have been inconclusive. Other agents implicated have been Yersinia enterocolitica and Borrelia burgdorferi, but the evidence linking these agents to sarcoidosis is even less well established. There is familial clustering of the disease, and sarcoidosis has been linked to human leukocyte antigens (HLA) A1 and B8 in whites, suggesting that hereditary and genetic factors play an important role in this disease. Non-MHC immunologic factors are probably also involved, as patients with sarcoidosis have numerous abnormalities of the immune system, including altered T-cell ratios, poorly responsive CD4+ T-cell subsets, hyperactive B-cell lines, and altered production of the inflammatory cytokines IFN-γ and RANTES by macrophages.23–27
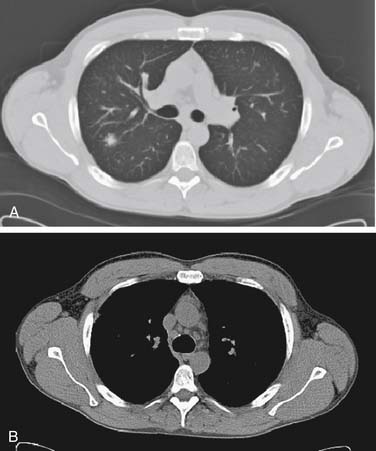
Symptoms of sarcoidosis are variable and fluctuate with time. Most patients in whom the disease is identified by routine chest radiography are asymptomatic. Others may present with the acute onset of constitutional symptoms such as fever, chills, fatigue, and weight loss. Afflicted individuals often have an associated connective tissue disorder, such as rheumatoid arthritis, systemic lupus erythematosus, and progressive systemic sclerosis. Pulmonary symptoms commonly include dyspnea and a dry cough.19 Diagnosis is typically made radiographically, with chest radiographs showing bilateral hilar and mediastinal lymph node enlargement with or without an accompanying reticulonodular pulmonary pattern indicative of interstitial disease. Consolidation and a ground-glass pattern may also be evident, particularly on chest CT (Fig. 11-3). Fibrosis is seen in later stages of the disease. Serum abnormalities may include an increase in levels of angiotensin-converting enzyme (ACE), presumably produced by the activated macrophages in the granulomas. A preponderance of lymphocytes in the BAL suggests an alveolitic process with granulomatous changes. Diagnosis, however, is most often made by mediastinal lymph node biopsy if adenopathy is present.19,28,29 In rare cases, transbronchial or VATS biopsy may be needed to establish the diagnosis. Histologic findings of affected pulmonary tissue show noncaseating granulomas with high tissue ACE levels.
Treatment consists primarily of steroids in symptomatic patients.19 Methotrexate and other immunosuppressive agents have also been used for refractory cases. Recent studies have suggested a benefit to tumor necrosis factor inhibitors such as infliximab in patients with poor responses to standard immunosuppressive regimens.30–32
Granulomatous Vasculitides: Wegener’s Granulomatosis and Churg-Strauss Syndrome
Like sarcoidosis, Wegener’s granulomatosis is a systemic disorder characterized by granuloma formation and it may involve multiple organ systems. Wegener’s is a rare disease with an incidence of 1 to 3 in 100,000. Affected individuals typically present between the ages of 40 and 60.33
In its most extreme form, Wegener’s granulomatosis is characterized by necrotizing granulomatous involvement of the pulmonary parenchyma and both pulmonary and renal vasculature.2,22,33–37 Disease expression is variable and may be limited to the pulmonary system. Numerous etiologies have been suggested, including infectious organisms such as parvovirus B19 and other respiratory agents, inhaled environmental agents such as silica, and genetic factors linked to the HLA alleles DR1, DR2, and DR12. Although all of these possible etiologies are supported by some evidence, no explanation is conclusive. The sera of individuals afflicted with the disease contain antibodies to antineutrophil cytoplasmic antibodies (ANCA). Specifically, 90% of patients with Wegener’s granulomatosis have C-ANCA, which chiefly binds to the PR3 plasma serine proteinase in neutrophils, as opposed to P-ANCA, which are antibodies to a perinuclear myeloperoxidase enzyme.22,34,35 The exact role of C-ANCA in the pathogenesis of Wegener’s granulomatosis is unclear and, unfortunately, ANCAs are also found in other disease states, such as systemic lupus erythematosus, in which ILD may develop.22,34–38
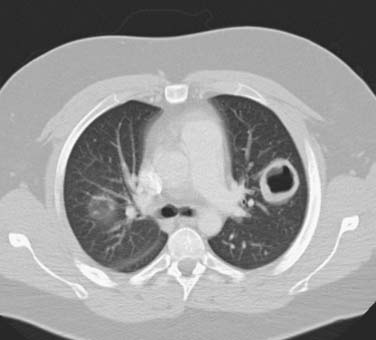
Individuals with Wegener’s granulomatosis may present with fulminant disease, with massive hemoptysis and renal failure secondary to extensive necrotizing granulomatous lesions, or with more subtle findings of fever, malaise, weight loss, and progressive dyspnea, with hemoptysis appearing only as the disease progresses. Neurologic or ocular symptoms may also be present. Diagnosis relies not only on the history but also on laboratory studies such as C-ANCA serologies and other associated systemic abnormalities.22,34,37 Chest imaging show a reticulonodular pattern consistent with interstitial disease or discrete nodules in only 50% of affected individuals. Central necrosis in these nodules can give the appearance of cavitary lesions (Fig. 11-4). BAL from patients with Wegener’s granulomatosis contains neutrophils and eosinophils. Biopsy of pulmonary or renal lesions may be necessary to establish or confirm the diagnosis. Pathology typically shows granulomas with neutrophils, macrophages, and eosinophils. Treatment options vary with the severity of the disease and include steroids and immunosuppressive agents, such as cyclophosphamide.37 Newer agents currently under active investigation include TNF inhibitors.4
Churg-Strauss syndrome is a rare systemic disorder characterized by eosinophilia, vasculitis, and pulmonary parenchymal granuloma formation. Many view this disease as part of a spectrum of disease that includes Wegener’s granulomatosis and other connective tissue diseases.22,35,36 The true incidence is unknown because of the difficulty of establishing a diagnosis, but it is estimated at 1 to 2 per 1 million people, and it affects individuals between the ages of 20 and 50.22,39 As in Wegener’s granulomatosis and sarcoidosis, multiple etiologies have been suggested, including pigeon exposure, helminth infection, and cocaine use. Recently, Churg-Strauss syndrome has been associated with the weaning of steroids in asthma patients placed on leukotriene antagonists.22,36,40,41
Affected individuals typically have a history of asthma or allergic symptoms and present with fever, cough, and occasionally hemoptysis. Gastrointestinal bleeding or neuropathy may also be present. Diagnosis rests on clinical suspicion and the combination of symptoms, laboratory studies, and pathologic findings. Chest radiographs typically reveal patchy consolidation throughout the lung fields. Levels of immunoglobulin E (IgE) and P-ANCA may be elevated. As expected, the BAL shows large numbers of eosinophils.28,29 Lung biopsy, usually required for diagnosis, demonstrates vasculitis and parenchymal granulomas with eosinophils. Treatment consists of steroid therapy and other immunosuppressive agents. Anti-TNF therapy has also been investigated.42 Prognosis for those diagnosed with the syndrome is poor, with a mortality of 50% to 60% at 5 years.35,36
Eosinophilic Pneumonias
Eosinophilic pneumonia belongs to a spectrum of pulmonary processes that are distinguished by the accumulation of eosinophils in parenchymal tissue. Eosinophilic pulmonary diseases with known etiologies include those caused by helminth infections, such as Strongyloides stercoralis and Ancylostoma duodenale, and drug allergies, which cause peripheral blood eosinophilia and can have systemic effects, as well as those conditions associated with the granulomatous vasculitides (discussed earlier) in which the etiology is unclear.43,44 All of these disease states can have pulmonary involvement. Diagnosis is made by history, blood eosinophil counts, blood serologies, and occasionally lung biopsy.
Idiopathic eosinophilic pneumonias can be divided into simple, acute, and chronic forms. Simple eosinophilic pneumonia, also known as Löeffler’s syndrome, is rare and characterized pathologically by interstitial edema with abundant eosinophils. Patients typically present with few or no symptoms. Chest radiographs show patchy parenchymal consolidation, and the diagnosis rests on the demonstration of peripheral blood eosinophilia in patients with a history of asthma or atopy. The disease usually resolves spontaneously, so biopsy or BAL is rarely required to make a diagnosis. Patients with symptoms, even when symptoms are prolonged, may benefit from steroid therapy.43,44
Acute eosinophilic pneumonia is a rare disorder that presents as an acute illness with severe respiratory distress that may require ventilatory assistance. The etiology is unclear, but it is thought to be secondary to an eosinophil-mediated immune response to an unknown allergen. Patients present with severe shortness of breath and pleuritic symptoms. Chest radiography show reticular opacities similar to the pattern found with interstitial pulmonary edema. The disease is characterized by a high number of eosinophils, up to 80%, in the BAL. Steroids are the mainstay of treatment, but a high relapse rate is associated with this disease.43,44
Chronic eosinophilic pneumonia has a similar histologic and, in most cases, radiographic appearance. Again, diagnosis relies on the demonstration of peripheral blood eosinophilia in patients with a history of asthma or atopy. However, affected patients have chronic symptoms that do not resolve over time, and therefore eosinophilia on BAL, tissue biopsy, or both, are often needed to make the diagnosis.29,44 Steroid treatment usually results in rapid disease regression.43,44
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


