A patient is classified as having SLE if he satisfies 4 of the clinical and immunologic criteria used in the SLICC classification criteria, including at least one clinical criterion and one immunologic criterion, OR if he or she has biopsy-proven nephritis compatible with SLE in the presence of ANAs or anti-dsDNA antibodies.
Criteria are cumulative and need not be present concurrently.
Clinical criteria
1. Acute cutaneous lupus, including:
Lupus malar rash (do not count if malar discoid)
Bullous lupus
Toxic epidermal necrolysis variant of SLE
Maculopapular lupus rash
Photosensitive lupus rash
In the absence of dermatomyositis
OR subacute cutaneous lupus
2. Chronic cutaneous lupus, including:
Classic discoid rash, localized or generalized
Hypertrophic (verrucous) lupus
Lupus panniculitis
Mucosal lupus
Lupus erythematosus tumidus
Chillblains lupus
Discoid lupus/lichen planus overlap
3. Oral ulcers (palate, buccal, tongue) OR nasal ulcers in the absence of other causes
4. Nonscarring alopecia (diffuse thinning or hair fragility with visible broken hairs)
5. Synovitis involving 2 or more joints
6. Serositis (Pleurisy or pericarditis)
7. Renal: urine protein-to-creatinine ratio (or 24-h urine protein) representing 500 mg protein/24 h OR red blood cell casts
8. Neurologic
Seizures
Psychosis
Moneuritis multiplex
Myelitis
Peripheral or cranial neuropathy
Acute confusional state
9. Hemolytic anemia
10. Leukopenia (<4,000/mm3 at least once) OR Lymphopenia (<1,000/mm3 at least once)
11. Thrombocytopenia (<100,000/mm3 at least once)
Immunologic criteria
1. ANA level above laboratory reference range
2. Anti-dsDNA antibody level above laboratory reference range (or >2-fold the reference range if tested by ELISA)
3. Presence of Anti-Sm antibody
4. Antiphospholipid antibody positivity as determined by any of the following
(a) Positive test result for lupus anticoagulant
(b) False-positive test result for rapid plasma regain
(c) Medium- or high-titer anticardiolipin antibody level (IgA, IgG or IgM)
(d) Positive test result for anti-β2-glycoprotein I (IgA, IgG or IgM)
5. Low complement
(a) Low C3
(b) Low C4
(c) Low CH50
6. Direct Coomb’s test in the absence of haemolytic anemia
The American College of Rheumatology classification criteria for systemic lupus erythematosus (SLE) were updated in 1997 [1] and again in 2012 [2]. Although lung involvement is not a criterion for SLE diagnosis, lung involvement has been associated with increased mortality [3, 4]. The prognosis of SLE greatly improved in the past years. The results from two prospective cohorts of 1,000 European [5] and 644 Canadian [3] patients with lupus found 95 and 93 % 5-year survival rates, respectively.
Interstitial lung disease in SLE may be acute or chronic. This review will focus on specific involvement of the lung by SLE, however it must be kept in mind that infection is the main cause of lung infiltrates in SLE. The risk of pulmonary infection is three time higher in patients with SLE than in the general population [6]. As infections are among the most important causes of morbidity and mortality in patients with SLE, an aggressive approach to SLE patients with new pulmonary infiltrates is mandatory, and infection should be presumed and treated empirically until an alternative diagnosis is given.
Acute Lupus Pneumonitis
Acute lupus pneumonitis (Fig. 26.1) occurs in 1–4 % of SLE patients. It often reveals a previously unknown SLE (50 % of the patients in the series of Matthay et al. [7] or may occur in the course of the disease.
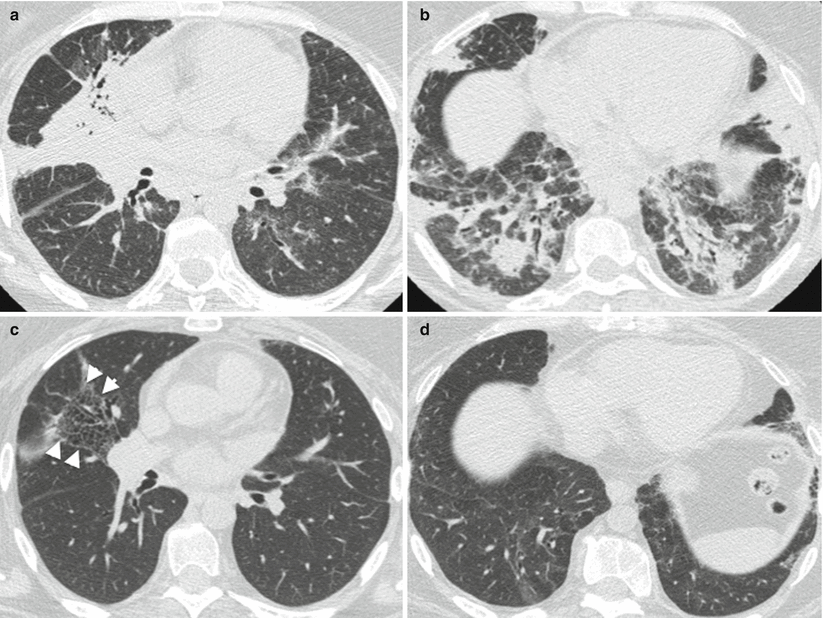
Fig. 26.1
Acute lupus pneumonitis revealed the systemic lupus erythematosus in a woman. High resolution computed tomography of the lungs showed areas of consolidation (panels a, b). After corticosteroid treatment, most of the opacities resolved (panels c, d). The right middle lobe was partly destroyed by residual fibrosis (panel d)
The clinical presentation is non specific, simulating an acute infectious pneumonia, with cough, dyspnea and fever. Hemoptysis is occasionally seen. Arterial blood gases analysis reveal hypoxemia with hypocapnia. Chest radiography and CT-scan show uni or bilateral alveolar infiltrates which usually predominate in the lower lobes. Small pleural effusions are commonly associated. Occasionally, acute respiratory failure, requiring mechanical ventilation will occur. A part from the rare occurrence of LE cells or the detection of hematoxylin eosin bodies, histological features obtained are non-specific and include alveolar wall damage and necrosis, alveolar edema, hyaline membranes, inflammatory cell infiltration and alveolar hemorrhage; capillary inflammation and thrombosis are also detected; deposits of immunoglobulins and complement are variably present [8, 9].
A syndrome of acute reversible hypoxemia with normal chest x-ray films, a normal CT scan and a rapid response to corticosteroids has been described in patients with SLE [10, 11]. The syndrome was attributed to leukoaggregation in the lung capillaries. Available histological data are very limited but demonstrate an infra-radiologic inflammation in the alveolar space [12]. This suggests that this syndrome is a less severe form of acute lupus pneumonitis rather than a distinct entity [12].
The clinicoradiographic presentation of lupus pneumonitis is absolutely non specific and may simulate lung infection, pulmonary embolism, or other acute pulmonary diseases. An invasive diagnostic workup must be set up and time is crucial since acute respiratory failure and death may develop. Bronchoalveolar lavage with a search for bacterial, viral, fungal and parasitic agents is required, but empirical antibiotherapy must not be delayed. CT-scan will appropriately characterize the lesions and exclude the potential diagnosis of pulmonary embolism. Lung biopsy has been advocated by some experts to exclude some diagnostics, however this procedure bears its own morbidity and mortality and lung histologic analysis is usually non diagnostic.
The treatment of acute lupus pneumonitis is based on case reports or small series. Empirical broad-spectrum antibiotic coverage should be maintained until infection is excluded. Mechanical ventilation and supportive intensive care may be required. The basis of drug therapy for acute lupus pneumonitis is high-dose corticosteroids (prednisone, 1–2 mg/kg/day) [13, 14]. Pulse methylprednisolone (250–1,000 mg/day for several days) have been used in patients with a severe initial presentation. Most patients will improve with this treatment despite 50 % mortality has been reported in older series [13, 14]. Cyclophosphamide, intravenous immunoglobulins, or plasma exchange may be used in cases of acute lupus pneumonitis refractory to corticosteroids. The place of new immunomodulatory agents such as anti-TNF has not been evaluated [15].
Diffuse Alveolar Hemorrhage
Diffuse alveolar hemorrhage is a rare but severe manifestation of SLE, most series reporting 50–90 % mortality [13, 16, 17] although a more favourable outcome has been reported in recent series [18, 19]. Its prevalence ranges between <2 and 5.4 % of patients with SLE.
The pathogenesis of the disease is poorly understood. Immune complex-mediated injury, vasculitis with alveolar capillaritis, alveolar damage related to infection, probably play a role. Acute inflammation and necrosis involving capillaries, arterioles, and small muscular arteries has been described [20]. The involvement of capillaries is manifested by an infiltrate of necrotic neutrophils within alveolar septa often associated with destruction of the alveolar wall. This capillaritis was present in all four cases originally described by Myers and Katzenstein [20], while involvement of arterioles and small arteries was seen in three. Immunofluorescence and electron microscopy demonstrated immune complexes in only two. In a recent series of 90 necropsies in patient s with SLE, capillaritis was observed in 3 of 23 patients with alveolar hemorrhage [21]. Capillaritis is not specific of SLE since it has also been described in alveolar hemorrhage associated with the antiphospholipid syndrome, in polymyositis and other connective tissue diseases, in Henoch-Schoenlein purpura, cryoglobulinemia, Behçet syndrome, and Wegener disease [22–24]. It may also be seen in antibasement membrane antibody disease.
Diffuse alveolar hemorrhage reveals SLE in 11–20 % of the cases [16]. Patients with lupus nephritis are at increased risk of developing alveolar hemorrhage, and renal involvement is observed in 60–93 % of the patients at diagnosis of diffuse alveolar hemorrhage. Microvascular renal and lung involvement appear to be pathogenetically similar [25]. The presentation ranges from asymptomatic to fulminant. Affected patients are young (mean age: 27 years) and present acutely ill with dyspnea, cough, fever and recent drop of hemoglobin blood levels [26]. Symptoms are usually abrupt in onset, being present for less than 3 days in two thirds of patients. Hemoptysis is initially present in less than half of patients. Bilateral lung infiltrates, ranging from limited ground glass opacities (Fig. 26.2) to dense consolidations are present. Arterial hypoxemia is common and more than 50 % of the patients will need mechanical ventilation [16].
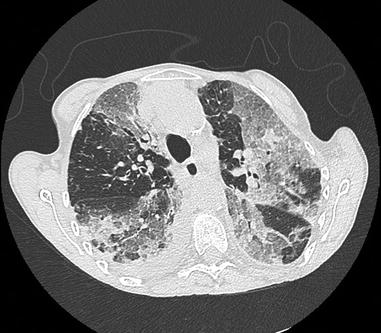
Fig. 26.2
Catastrophic antiphospholipid syndrome in a woman with longstanding systemic lupus erythematosus and antiphospholipid syndrome. No trigger was identified. CT scan shows multiple well limited ground glass opacities with some areas of consolidation, with a right pleural effusion. Bronchoalveolar lavage fluid analysis showed a neutrophilic alveolitis. Partial improvement was obtained with corticosteroids
The diagnosis of alveolar hemorrhage is usually easily obtained with bronchoalveolar lavage, which allows for a search for infectious agents. Concomitant lung infection, bacterial, fungal or viral, is observed in about one third of patients, and bears a poor prognosis [16]. Lung biopsy, either transbronchial or surgical, is not useful once the diagnosis of SLE is ascertained with the presence of antinuclear antibodies, and may be dangerous in critically ill patients. Echocardiography is mandatory to evaluate the presence of valvular or myocardial dysfunction. The classical increase of DLCO observed at the early stages of alveolar hemorrhage is of limited interest in critically ill patients [27].
The treatment of SLE-associated alveolar hemorrhage is not well defined. High dose corticosteroids are commonly used (methylprednisolone 1 g daily for 3 days, progressively decreased over the next days) but corticosteroids alone does not appear to be very effective. In a recent series, diffuse alveolar hemorrhage developed in patients already treated with high dose corticosteroids for lupus nephritis [17]. A combination of corticosteroids, cyclophosphamide and plasmapheresis has been used with promising results [28, 29]. Plasmapheresis should be reserved for patients with severe alveolar hemorrhage refractory to corticosteroids and cyclophosphamide [30]. A systematic search of respiratory infections and the addition of broad-spectrum antibiotics for the initial treatment of lupus patients with diffuse alveolar hemorrhage is recommended based on the lower mortality reported in some studies [19, 31]. Survivors are exposed to the risk of developing pulmonary fibrosis [32]. Diffuse alveolar hemorrhage may recur several times in the same patient but the factors associated with recurrence are not well understood. In a recent series from Mexico, an increased risk of death was associated with thrombocytopenia, renal failure, requirement for mechanical ventilation and high severity score as assessed by the Acute Physiology And Chronic Health Evaluation II score (APACHE II) [33].
Acute Respiratory Distress Syndrome
The prevalence of adult respiratory distress syndrome (ARDS) ranges from 3.5 to 15 % of patients with lupus [34]. A retrospective study of 544 Korean lupus patients found 19 (3.5 %) cases of ARDS with a mortality rate of about 70 % [34]. Death related to ARDS was found in one third of all deaths from lupus during the 4-year study period. The most frequent cause of ARDS was sepsis or bacteraemia (47.4 %). The main organisms causing the sepsis were gram-negative bacilli (61.5 %). The ARDS developed at a younger age and progressed more rapidly than ARDS in non-SLE patients.
Catastrophic Antiphospholipid Syndrome
The catastrophic antiphospholipid syndrome is a rare and excessively severe manifestation of the antiphospholipid syndrome which is observed both in primary and secondary antiphospholipid syndrome [35] (Fig. 26.2). The syndrome is characterized by multiple simultaneous vascular occlusions throughout the body. The lung is involved in 66 % of the cases, with ARDS, pulmonary embolism, pulmonary artery thrombosis, pulmonary microthrombi, or alveolar hemorrhage, sometimes associated [36]. About 50 % of the patients die with the syndrome. Lung involvement and presence of SLE is associated with a twofold increased risk of death [37]. With an adequate treatment, including high dose steroids, anticoagulation, plasma exchanges or intravenous immunoglobulins, the prognosis seems better, and a mortality below 10 % is observed in experienced groups [38]. Long term anticoagulation is needed after recovery, as well as the treatment of SLE.
Chronic Interstitial Lung Disease
Clinically significant chronic interstitial pneumonia rarely complicates SLE and extensive lung fibrosis is rarely observed (3 % of the patients in one study) (Figs. 26.3 and 26.4) [39]. An autopsy study of 120 patients with lupus revealed a prevalence of interstitial pneumonitis or fibrosis of about 13 % [8]. However systematic CT evaluation of non selected patients with SLE demonstrated the high prevalence of subclinical interstitial abnormalities, observed in 30–40 % of the patients [40]. Detected abnormalities consist mainly of thickened inter-lobular walls, of limited extension, and usually predominant in the subpleural regions. Pulmonary function tests were abnormal in about 50 % of the patients with abnormal HRCT, but HRCT changes did not correlate with pulmonary function abnormalities [40]. In one series, however, the extent of disease was statistically significantly correlated with duration of clinical history and decreased single-breath diffusing capacity for carbon monoxide and ratio of forced expiratory volume in 1 s to forced vital capacity [40].
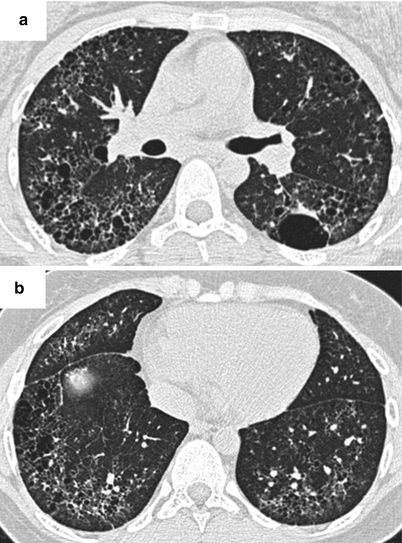
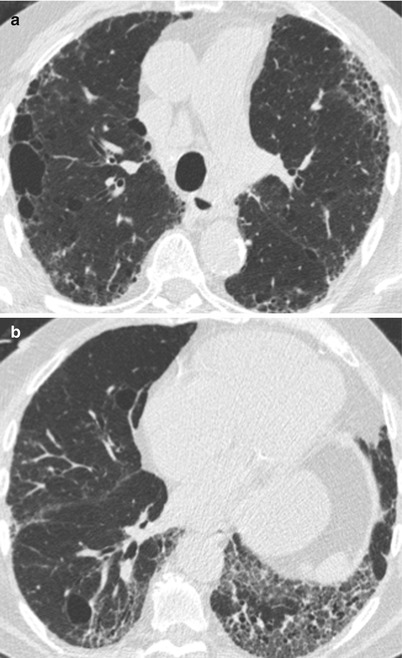
Fig. 26.3
(a, b) Lung fibrosis in a patient with systemic lupus erythematosus. Cysts of different size are observed, associated with reticulations
Fig. 26.4
(a, b) Combined pulmonary fibrosis and emphysema syndrome in a patient with systemic lupus erythematosus
The larger series of Eisenberg and colleagues described 18 patients, identified over a 1-year period, with radiographic evidence of pulmonary fibrosis, representing less than 3 % of the patients followed at their institution [39]. All the patients had a restrictive functional pattern but only seven were symptomatic. In most cases, chronic ILD develops insidiously, sometimes with mild flares of lung involvement [39]. In some patients, lung fibrosis could be the sequela of acute pneumonitis. Lung involvement does not correlate with any biological characteristic, although an association between anti-SS-A antibodies or anti-U1 RNP antibodies and chronic interstitial pneumonia was observed [41, 42].
Histologic reports describe non specific abnormalities with interstitial lymphocytic infiltrates, interstitial fibrosis, and honeycomb changes, sometimes associated with follicular bronchiolitis [39, 43]. Non specific interstitial pneumonia (NSIP) is likely the most common pattern in patients with pulmonary fibrosis, although the real incidence of NSIP in SLE is still not well defined. Desquamative interstitial pneumonia has been reported [44]. Long term evolution of chronic ILD in SLE is poorly known and treatment is not evaluated. In the Weinrib’s series, all patients were treated with corticosteroids, and the condition improved in 9/14 [39]. Improvement with oral methotrexate, mycophenolate mofetil or UVA has been reported [45].
Lymphocytic interstitial pneumonia (LIP) has been described in a few patients with SLE, usually in association with Sjögren’s syndrome [46, 47]. In these cases, the development of lung cysts should suggest the diagnosis of LIP [47]. Finally, the clinicoradiologic syndrome of organizing pneumonia (formerly known as BOOP) characterized by patchy alveolar infiltrates and an histologic pattern of organizing pneumonia has been described in patients with SLE (Fig. 26.5). These cases usually demonstrate a rapid improvement with corticosteroids [48]. The concurrence of sarcoidosis and SLE has been reported in a few cases [49]. Nodular amyloidosis, excavating nodules, have also been observed.
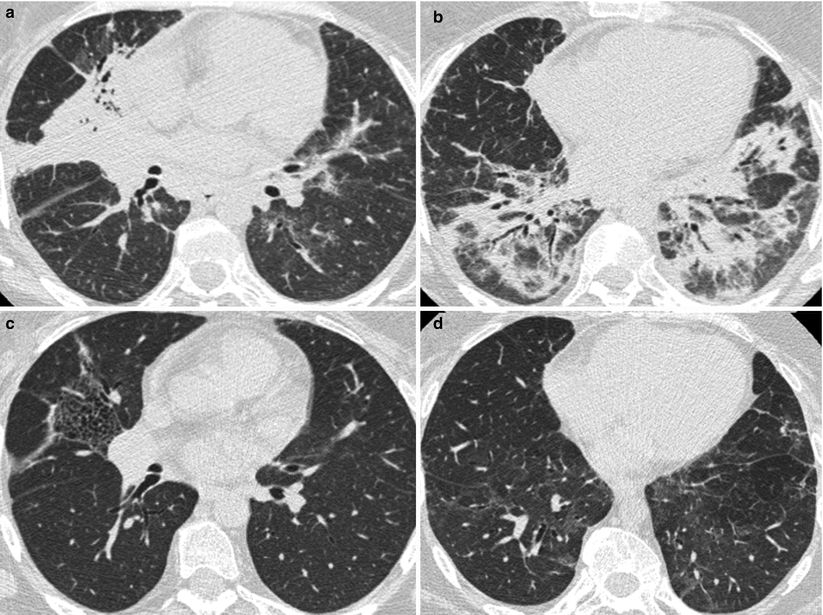
Fig. 26.5
Organizing pneumonia revealing systemic lupus erythematosus. Bilateral consolidations (panels a, b) resoved with corticosteroids (panels c, d)
The Shrinking Lung Syndrome
The term “shrinking lung syndrome” has been applied to SLE patients presenting with progressive dyspnea, the characteristic chest radiographic findings of small lung volumes, elevated hemidiaphragms and bibasilar infiltrates, with a restrictive ventilatory defect and a preserved carbon monoxide transfer coefficient [50] (Fig. 26.6). Pleuritic pain is also a frequent symptom in these patients [51]. Shrinking lung syndrome can be present in any phase of the disease and can even be its first manifestation. This syndrome was attributed to diaphragmatic dysfunction on the basis of the demonstration of decreased inspiratory muscle strength in 11 SLE patients [52]. Conversely, Laroche et al., using bilateral electrostimulation in 12 patients with the shrinking lung syndrome, failed to demonstrate diaphragm weakness [53]. In a well documented case, Hardy described a patient with the syndrome and bilateral phrenic nerve paralysis [54]. With corticosteroids, the phrenic nerve function recovered whereas the restrictive functional pattern persisted, suggesting that reduced diaphragm muscle contractility per se does not explain the small volume lungs and respiratory symptoms in patients with the syndrome. Hawkins reached similar conclusions in a different patient [55]. However, lupus myopathy has been associated with the syndrome [56]. We suggest that recognition of this syndrome requires a thorough evaluation to determine its cause and give the adequate treatment.
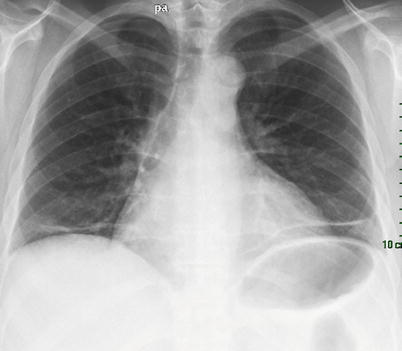
Fig. 26.6
Shrinking lung syndrome in a patient with systemic lupus erythematosus. Note the rise of diaphragm and the linear basal opacities
Some improvement of dyspnea and restriction has been observed with corticosteroids [50, 57]. Anecdotal data support the use of theophyllin, beta-2-agonists, cyclophosphamide, azathioprine, or rituximab, in patients unresponsive to steroids [58, 59]. Patients seem to stabilize and have no worsening of lung function with time.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is the most common connective tissue disease (see Table 26.2 for classification criteria). As is the case for SLE, the pleuropulmonary manifestations of RA are varied, pleural abnormalities and ILD being the more common. Although RA affects women preferentially, men are more affected by pleuropulmonary manifestations of the disease. The diagnosis of RA is helped by serological markers, rheumatoid factor and anti-cyclic citrullinated peptide antibodies which are included in the 2010 rheumatoid arthritis classification criteria [60]. Tobacco-smoking is a risk factor for the development of ILD in patients with RA and is a strongly associated environmental risk factor for RA. There is a strong association between cigarette smoking and the presence and titre of rheumatoid factors [61, 62]. Although not replicated in all cohorts, recent studies demonstrate that the increased risk conferred by tobacco exposure is restricted to the anti-cyclic citrullinated peptide antibodies -positive subset of subjects and is associated with HLA-DR alleles [63, 64]. Analysis of lung samples from smokers shows citrullination of cellular proteins that is not present in nonsmoking subjects thereby providing a source of citrullinated antigens to induce autoantibody production [65]. There is evidence of RA-specific antibodies in patients wit ILD without RA [66]. The level of anti-cyclic citrullinated peptide antibodies are increased in patients with RA with exposure to tobacco [67].
Table 26.2
American College of Rheumatology/European League against Rheumatism Classification criteria for Rheumatoid arthritis [200]
The criteria apply to patients with at least one clinical synovitis not better explained by another diagnosis. |
Score of ≥6 points: |
1. Joint involvement |
(a) 1 large joint (0 points) |
(b) 2–10 large joints (1 point) |
(c) 1–3 small joints (2 points) |
(d) 4–10 small joints (3 points) |
(e) More than 10 joints (at least one small) (5 points) |
2. Serology |
(a) Negative rheumatoid factor (RF) and negative anticyclic citrullinated peptide antibody (aCCP) (0 points) |
(b) Low positive RF or aCCP (≤3 times the upper limit of normal (2 points) |
(c) High positive RF or aCCP (>3 times the upper limit of normal (3 points) |
3. Acute phase reactants |
(a) Normal C-reactive protein and normal erythrocyte sedimentation rate (0 points) |
(b) Abnormal C-reactive protein or abnormal erythrocyte sedimentation rate (1 point) |
4. Duration of symptoms |
(a) <6 weeks (0 points) |
(b) ≥6 weeks (1 point) |
Frequency of Interstitial Lung Disease
Interstitial lung disease is the predominant pulmonary manifestation of RA. ILD is usually detected in patients already diagnosed with RA (mostly between the ages of 50 and 60 year), but isolated pulmonary disease may precede the onset of articular disease [68]. Clinically significant RA-ILD affects 7–10 % of patients with RA [68–70], whereas a higher prevalence was found by autopsy studies (interstitial changes are observed in 80 % of lung biopsies) or prospective screening studies using high-resolution chest tomography (up to 50 %) or lung function tests (a decrease of the diffusing capacity of carbon monoxide is observed in up to 40 % of RA patients) [71, 72]. In a population-based incidence cohort of patients with RA and a matched cohort of individuals without RA, the hazard ratio (HR) of developing ILD in RA patients was 8.96 (95 % confidence interval [95 % CI] 4.02–19.94) as compared with controls [68]. The risk of developing ILD was higher in RA patients who were older at the time of disease onset, in male patients, and in individuals with more severe RA [68]. The risk of death for RA patients with ILD was three times higher than in RA patients without ILD (HR 2.86 [95 % CI 1.98–4.12]). Median survival after ILD diagnosis was only 2.6 years. In that study, ILD contributed approximately 13 % to the excess mortality of RA patients when compared with the general population. Whereas overall mortality rates for RA have fallen, those associated with RA-ILD have increased significantly in older age groups [70]. The presence of ILD was associated with a reduction of life expectancy of about 2 years [70, 73]. Asymptomatic preclinical ILD, which is detectable by HRCT, may be prevalent (up to 35 % of patients without respiratory symptoms) and progressive (in about half the patients in one series) among patients having RA [74–76]. Cigarette smoking seems to be associated with preclinical ILD in patients having RA, and treatment using methotrexate may be a risk factor for progression of preclinical ILD [75]. The influence of disease-modifying antirhumatic drugs on the development and evolution of RA-ILD is unknown. Recent data from the British data base suggest that the mortality associated with RA-ILD is not increased by antiTNF agents [77].
Histopathological Patterns
Histopathological findings in RA-associated ILD disclose very different patterns, sometimes associated : Usual interstitial pneumonia (UIP), NSIP, Desquamative interstitial pneumonia, LIP, organizing pneumonia, eosinophilic infiltration [9]. Many cases are difficult to classify into one pattern. Follicular bronchiolitis consisting of lymphoid hyperplasia and reactive germinal centers along small airways is also detected, associated or not with a LIP pattern [78]. The relative prevalence of each histopathological pattern varies according to the studies, however the currently available data indicate that the histopathological pattern of UIP may be equally as frequent as NSIP in patients with RA-ILD, contrasting with the predominance of NSIP in the other connective tissue diseases [79]. However, UIP in connective tissue disease may not exactly replicate idiopathic UIP (as in idiopathic pulmonary fibrosis), with fewer fibroblastic foci [80, 81], smaller honeycombing spaces, and more pronounced inflammation and germinal centres [81]. A trend was found toward worse survival of RA-ILD with UIP histological pattern as compared to those with a NSIP pattern [82].
Radiographical Patterns
The most common CT features of RA-ILD are ground glass opacities (90 % of patients) and reticulations (98 % of patients) [83]. In that study, four major CT patterns were identified: UIP (n = 26) (Fig. 26.7), NSIP (n = 19) (Fig. 26.8), bronchiolitis (n = 11), and organizing pneumonia (n = 5). There was a good concordance between the HRCT pattern and the histopathological pattern [83].
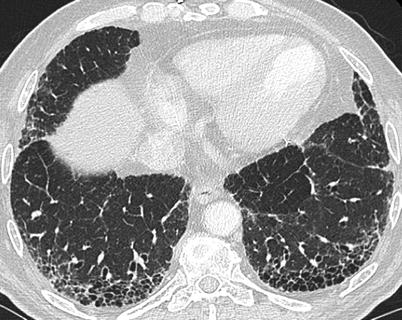
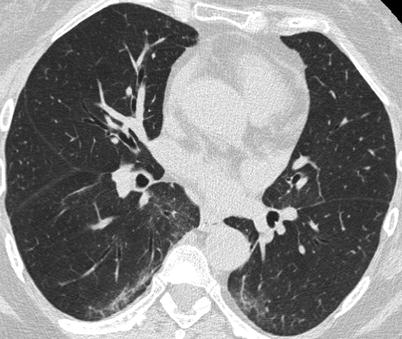
Fig. 26.7
A usual interstitial pneumonia (UIP) pattern in a patient with rheumatoid arthritis
Fig. 26.8
A non specific interstitial pneumonia (NSIP) pattern in a patient with rheumatoid arthritis
The prognosis of RA-associated ILD is usually good as the deterioration of lung function is slow [74, 75]. However one study reported a median survival of 3.5 years and a 5-year survival rate of 39 % in 49 patients with RA hospitalized for interstitial pulmonary fibrosis, a survival very similar to what is observed in patients with idiopathic pulmonary fibrosis [84]. The prognosis of RA-ILD is better assessed by the HRCT pattern. In a recent study based on HRCT, a definite UIP pattern was seen in 20 (24 %) out of 82 patients with RA-ILD. These patients showed worse survival than those without this pattern (median survival 3.2 versus 6.6 years), and a similar survival to those with idiopathic pulmonary fibrosis [85]. On multivariate analysis, a definite UIP pattern on HRCT was associated with worse survival (hazard ratio of 2.3). Analysis of specific HRCT features demonstrated that traction bronchiectasis and honeycomb fibrosis were associated with worse survival (hazard ratio of 2.6 and 2.1, respectively). Female sex (hazard ratio of 0.30) and a higher baseline diffusing capacity of the lung for carbon monoxide (hazard ratio of 0.96) were associated with better survival [85]. This was confirmed by Tsuchiya and colleagues who observed that the 5-years survival rates were 36.6 % in the UIP group and 93.8 % in the NSIP group [86]. These studies suggest that a pragmatic imaging-based approach is possible and routinely applicable to approach the prognosis in RA-associated ILD [87], although the case of patients with undefinite HRCT pattern remains uncertain [85].
Treatment of RA-ILD
Treatment of RA-ILD is not well defined. It should be tailored according to the pattern of lung involvement. If organizing pneumonia or NSIP patterns predominate, with ground glass and consolidations, corticosteroids are first-line therapy and may be effective alone, but most often an immunosuppressant will be needed. If the HRCT pattern is uncertain, a combination of steroids and immunosuppressants may be used, over a limited period of time and maintained if an objective improvement of lung function tests is obtained. In case of a UIP pattern, some still consider a combination of steroids and immunosuppressants [88]. Cyclophosphamide, azathioprine, methotrexate, cyclosporine, have been used. At this time, the best treatment regimen is not defined. Improvement of lung involvement has been reported in one case with blockade of interleukin-6 receptor [89].
Acute Exacerbation of RA-ILD
An ILD exacerbation is generally defined as rapidly deteriorating respiratory symptoms within a 30-day period with evidence of new infiltrates (usually new ground glass opacities) and exclusion of an identifiable cause. The absolute risk of an acute exacerbation is not well established, but based on the limited data of acute exacerbations in collagen vascular diseases, possibly as many as 20 % of patients with RA-ILD will experience an acute exacerbation with a 1-year incidence as high as 2.58 % [90]. By the time patients present to the hospital, hypoxemia may be severe and a ventilatory support may be needed.
The diagnosis of exacerbation requires the exclusion of alternative explanations for a clinical worsening, such as infection, drug reaction, left ventricular failure or pulmonary embolism. Treatment usually associates methylprednisolone pulses and immunosuppressants (IV cyclophosphamide) [90, 91]. The prognosis is very poor [90, 91].
Rheumatoid Nodules
Rheumatoid nodules are the only specific lesion observed in the lung of RA patients. Rheumatoid nodules are histologically similar to that observed in the subcutaneous tissue. Occasionally, giant cells and well-formed granulomas may be observed in the peripheral region of the nodule [72]. Very frequent at microscopic examination of the lung (30 %), or on HRCT lung slices (20 %), nodules are seldom seen on standard chest X ray (<1 %). Nodules usually predominate in the upper and mid-lung regions, in the peripheral sub-pleural zone, although endobronchial nodules do exist. The nodules are more prevalent in smokers, in males, and in patients with extra-articular manifestations or with subcutaneous nodules [92]. Multiple widespread nodules have been described as rheumatoid nodulosis and may be induced by treatment such as methotrexate, leflunomide or antiTNF agents [93–95]. Nodules are usually asymptomatic and do not evolve over time, but cavitation and infection may occur [96]. Detection of one or more lung nodules in a patient with RA poses the problem of their nature. A systematic diagnostic workup is needed in order not to miss an infectious or tumoral lesion. Increased fluorine-18-fluorodeoxyglucose uptake may be misleading [97].
A syndrome of bilateral lung nodules in silica-exposed RA patients has been described as the Caplan’s syndrome, also observed in other dust exposed RA patients [98]. The histopathological image of the nodules is similar to the rheumatoid nodule except for the presence of an additional peripheral pigmented dust surrounding the lesion [72]. Most patients have a pre-existing mild pneumoconiosis.
Other Patterns
Combined pulmonary fibrosis and emphysema was described in 18 RA patients [99] (Fig. 26.9). Patients with combined pulmonary fibrosis and emphysema had higher lung volumes, lower diffusion capacity, higher pulmonary artery pressures, and more frequently were male than those with connective tissue diseases and lung fibrosis without emphysema [99]. Secondary amyloidosis involving the lung with an interstitial pattern is a rare but possible complication of long lasting RA [100]. Apical fibrobullous disease, similar to what is observed in patients with ankylosing spondylarthritis, is observed in RA patients [101].
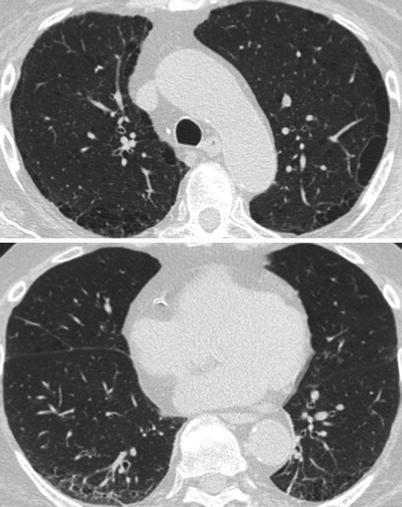
Fig. 26.9
Combined pulmonary fibrosis and emphysema syndrome in a patient with rheumatoid arthritis. Paraseptal emphysema is observed in the upperlobes (upper panel) while subpleural reticular opacities are evident in the lower lobes (lower panel)
Alveolar haemorrhage related to pulmonary vasculitis has been reported [102], sometimes with antineutrophil cytoplasmic antibodies [103] (Fig. 26.10).
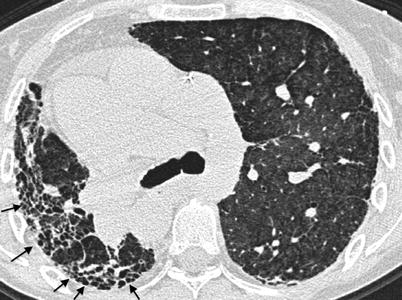
Fig. 26.10
A woman with rheumatoid arthritis and lung fibrosis with a right lung predominance (typical honeycombing with loss of volume is present in the right lung) developed an acute alveolar hemorrhage with diffuse ground glass opacities in the left lung. ANCA-associated vasculitis involving the lung and the kidney was diagnosed. Arrows: typical honeycombing in the right lung
Drug-Induced Lung Disease
Almost all drugs used for the treatment of RA have been associated with drug-induced lung disease. Undesirable respiratory side-effects of methotrexate, gold salts, D-Penicillamine, and nonsteroidal anti-inflammatory drugs are very well described. However, new compounds (such as anti-TNF agents, sirolimus, leflunomide, tocilizumab, rituximab), and new clinicoradiological patterns are continuously described [77, 104–107]. Reference to available comprehensive reviews [108] and to Pneumotox®, an internet searchable database which gives up-to-date information for this rapidly evolving field are needed in order to determine whether any respiratory abnormality in a RA patient could be secondary to the treatment received. There could be a genetic predisposition to drug-induced pneumonitis [109], as suggested by the observation that the incidence of leflunomide-associated pneumonitis is 0.1 % in western populations and 1 % in Asian populations [110, 111]. Interestingly, a preexisting ILD increases the risk of developing a drug-induced pneumonia, as shown with methotrexate, leflunomide or anti-TNF molecules [106, 110–112]. For instance, a preexisting ILD increases 7-fold the risk of methotrexate-associated pneumonitis [112], and eightfold the risk of leflunomide-associated pneumonitis [111]. This suggests that the lung is primed to inflammatory reaction in patients with RA.
A preexisting ILD is usually not a contraindication to the use of MTX, leflunomide or anti-TNF agents in patients with rheumatoid arthritis, since these drugs are very useful for the patients, and the basal risk of pneumonia development in a given patient remains very low, even in case of preexisting ILD. However, the respiratory consequences of a drug-induced pneumonitis will be worst in case of preexisting ILD and mortality associated with drug-induced ILD is increased twofold in patients with underlying ILD [106]. Some experts recommend that patients with RA starting methotrexate undergo a baseline chest radiograph and pulmonary function tests, which can be used for comparison if respiratory symptoms develop [113], although this has not been incorporated into formal guidelines. The 2008 recommendations on RA treatment issued by the American College of Rheumatology stated that methotrexate was contraindicated in the presence of clinically important RA-associated pneumonitis or interstitial lung disease of unknown cause [114]. This recommendation was not amended in the 2012 updated recommendations [115]. No definition of a clinically important ILD was given in that recommendation, but we may suggest that methotrexate should be avoided in a patient with RA-associated ILD responsible for chronic respiratory insufficiency requiring oxygen therapy. If methotrexate use is deemed hazardous, one may consider using preferentially rituximab (anti-CD20) since the lung-toxicity of this drug was essentially described outside of the rheumatology field [107, 116], instead of leflunomide or anti-TNF agents.
Sjögren’s Syndrome
The Sjögren’s syndrome (SS) is one of the most common autoimmune diseases, characterized by the infiltration of different organs by CD4-positive T lymphocytes, the lacrymal and salivary glands being the most often involved (see Table 26.3 for classification criteria). The classic triad associates xerostomia (dry mouth), xerophtalmia (dry eyes) and arthritis. Multiple diagnosis criteria have been proposed, and have been recently updated [117]. Criteria now require the presence of either an anti-SSA or anti-SSB autoantibody, or a typical lesion on the accessory gland biopsy (Chisholm grade 3 or 4). Detection of an anti-SSA antibody in a patient with idiopathic ILD is rarely indicative of a SS [118]. SS may be isolated (primary SS) or associated with a definite connective tissue disease (secondary SS, primarily with RA). These criteria have a specificity of 97 % and a sensitivity of 90 % for the diagnosis of secondary SS. Lung involvement is less common and less severe in primary SS than in secondary SS.
Table 26.3
Classification criteria for Sjögren’s syndrome
SICCA (Sjögren’s International Collaborative Clinical Alliance Cohort) 2012 criteria [201] |
|---|
At least 2 of the following 3 objective features: |
1. Positive serum anti-SSA/Ro and/or anti-SSB/La or (positive rheumatoid factor and ANA titer ≥1:320) |
2. Labial salivary gland biopsy exhibiting focal lymphocytic sialadenitis with a focus score ≥1 focus/4 mm2 (Chisholm grade ≥3) [202] |
3. Keratoconjunctivitis sicca with ocular staining score ≥3 (assuming that individual is not currently using daily eye drops for glaucoma and has not had corneal surgery or cosmetic eyelid surgery in the last 5 years) [203] |
Prior diagnosis of any of the following conditions would exclude participation in SS studies or therapeutic trials because of overlapping clinical features or interference with criteria tests: |
History of head and neck radiation treatment; Hepatitis C infection; acquired immunodeficiency syndrome; sarcoidosis; amyloidosis; graft versus host disease; IgG4-related disease |
Prevalence of Interstitial Lung Disease
The reported prevalence of pulmonary disease in SS varies widely according to the diagnostic modalities used to identify the abnormalities. HRCT detects abnormalities in 34–65 % of SS patients evaluated [119–123]. A comprehensive evaluation including lung function tests detected abnormalities in 75 % of SS patients [124]. However, if one considers only clinically significant pulmonary disease, it is estimated to affect less than 10 % of SS patients although a 22 % prevalence has been recently reported [125, 126]. Airways involvement and ILD are the most frequent manifestations of lung involvement in SS.
Interstitial lung disease is common in patients with SS and may reveal the disease. It affects 8–38 % of patients with primary SS. Bronchoalveolar lavage has demonstrated the high prevalence of subclinical lymphocytic and neutrophilic alveolitis, affecting 50 % of SS patients [127]. Alveolitis is more frequent in patients with extra-pulmonary involvement. An expansion of CD8+ T-lymphocytes has been associated with more frequent alteration of lung function tests [128].
Histopathology
The histopathology of ILD in SS is not specific. Different histological patterns may be observed, sometimes associated in a given individual: desquamative interstitial pneumonia, NSIP, UIP, LIP, organizing pneumonia, end stage lung [9, 129–131]. Bronchiolitis is frequently associated. NSIP is the more common pattern [130, 131]. Well formed granulomas may be seen in up to 10 % of samples [129].
Clinical Patterns
As in other connective tissue diseases, various radioclinical presentations are possible, and can be classified according to the chronology of involvement.
Acute or subacute infiltrating pneumonia, developing over several days or a few weeks may be due to organizing pneumonia [132] (Fig. 26.11), acute NSIP, eosinophilic pneumonia [133] or sometimes an exacerbation of a previously unrecognized diffuse infiltrative pneumonia [134]. This may be the first presentation of Sjögren’s syndrome. In this setting, alveolar haemorrhage is very rare and requires the search for cryoglobulinaemia [135] or an associated connective tissue disease, mainly systemic lupus erythematosus. Organizing pneumonia in the context of SS respond quite well to corticosteroids.
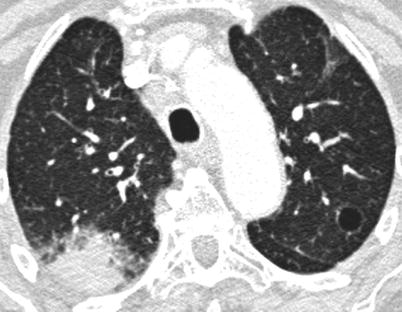
Fig. 26.11
Acute organizing pneumonia in a woman with a primary Sjögren’s syndrome. Note the cyst of the upper left lobe
Chronic ILD, developing over several weeks or several months.
This type of involvement can reveal SS. The radiographic appearance can suggest NSIP with ground glass opacities, mainly subpleural and posterior, with signs of fibrosis variable in extent (subpleural reticulations, sometimes honeycomb cysts) (Fig. 26.12). Disease progression is often very slow, over many years. It is often difficult to differentiate from LIP in the absence of a surgical lung biopsy. In some cases, imaging is suggestive of UIP with honeycomb cysts peripheral and subpleural in distribution (Fig. 26.13).
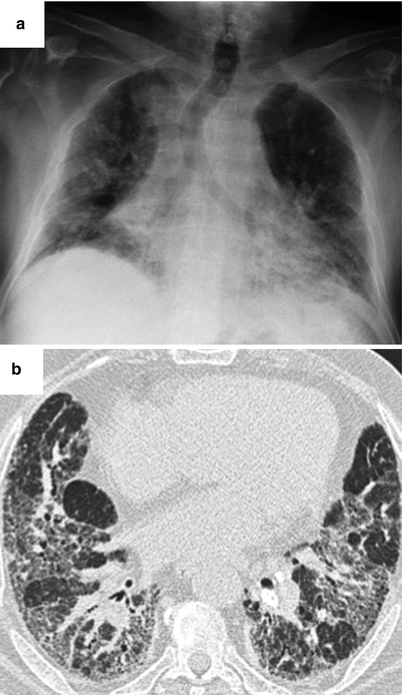
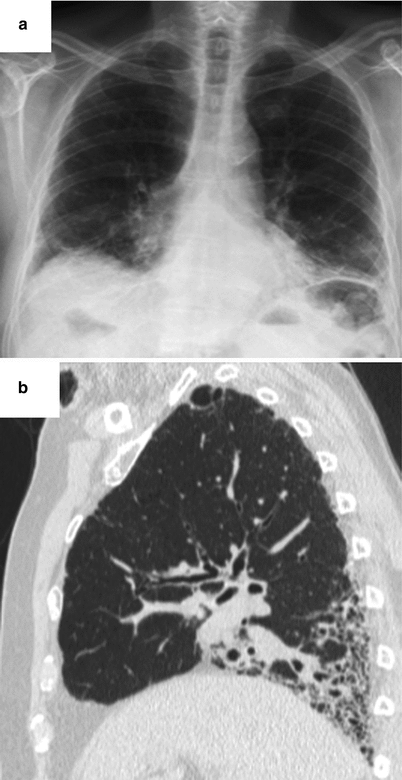
Fig. 26.12
Lung fibrosis with a fibrotic NSIP pattern revealed the primary Sjögren’s syndrome. Chest X ray (panel a) and CT scan (panel b)
Fig. 26.13
Lung fibrosis with UIP pattern in a patient with primary Sjögren’s syndrome. Chest X ray (panel a) and CT scan (panel b)
Information concerning the evolution and the treatment of ILD in SS are limited. Available data suggest that ILD in primary SS has a good prognosis without evidence of clinically significant deterioration over time in most patients [125, 136–138], although progression of lung disease with development of chronic respiratory failure may occur, particularly in patients with a UIP pattern [138]. In a recent series, 5 years survival rate of patients with SS and ILD was 84 % [137]. Ten of thirty-three patients died in the course of the follow-up period: five with fibrosing NSIP, two with malignant lymphoma, one with chronic bronchiolitis, two with fibrosis. Nine of the ten patients died of causes related to the disease. Six died of progressive respiratory failure independent of efficacy of initial treatment [137].
The treatment of ILD in patients with Sjögren’s syndrome is not well defined [139]. Hydroxycholoroquine has been shown to reduce sicca symptoms in a retrospective study [140] but its effect on pulmonary involvement was not evaluated. Deheinzelin reported the evolution of 11 patients treated with azathioprine alone or combined with prednisone [129]. The condition of seven patients improved (symptomatic relief and increase of vital capacity) and one patient deteriorated. Among five untreated patients, only one improved. The respective position of these treatments is not clear but a trial of prednisone and immunosuppressants should be performed in symptomatic patients. Improvement of ILD with rituximab has been reported in six out of eight patients treated in one series [141], but the characteristics of lung involvement were not precisely described in that study.
Lymphocytic Interstitial Pneumonia
With follicular bronchiolitis and BAL lymphocytosis, LIP is one of the pulmonary lymphoproliferative disorders associated with SS. LIP is a rare infiltrating pulmonary disorder. The definitive diagnosis requires a lung biopsy showing lymphoplasmocytic infiltration of the interstitial tissue and the alveolar septa, associated with lymphoid follicles and sometimes germinal centres [142]. Epithelioid and giant cell granulomas, and limited areas of organizing pneumonia, can be observed [142]. A coexistence with follicular bronchiolitis is possible; the final histopathological diagnosis will depend on the dominant component of the lesion. Histological diagnosis of LIP is sometimes difficult [142]. LIP must be differentiated from other lymphoid pulmonary infiltrations, mainly MALT (Mucosa Associated Lymphoid Tissue) lymphoma; this requires the use of immunohistochemical techniques (lymphocyte immunophenotyping) and molecular biology techniques (study of immunoglobulin gene rearrangements) to identify a clonal lymphocyte population. Other pulmonary disorders can be mistaken for LIP, in particular hypersensitivity pneumonitis or cellular NSIP. Sjögren’s syndrome must be sought systematically, as SS is the cause of LIP in 25–50 % of cases [142–144].
The CT scan occasionally suggests the diagnosis when it shows centrilobular and subpleural nodules associated with ground glass opacities, and thickening of peribronchovascular tissues and the interlobular septa. Parenchymal cysts are present in more than one in two cases, and enlarged mediastinal lymph nodes variable in size are present in one case in two [143]. Limited areas of pulmonary consolidation are possible [143]. Few data are available concerning the evolution of LIP. In a study of 14 patients followed-up for 13 months, 9 patients improved, and 1 was stable, while 4 patients worsened and developed fibrotic lesions with honeycombing [145]. In another series, progression to respiratory failure was observed in 3 of the 15 patients studied (20 %) despite treatment [144]. The issue of a possible evolution of LIP to lymphoma has not been entirely settled. Some observations in the literature could correspond to undiagnosed MALT lymphomas.
The treatment of LIP is not well established. Corticosteroids are usually administered, sometimes associated with immunosuppressants (methotrexate, cyclophosphamide, azathioprine, cyclosporin) [144]. New treatments targeting B lymphocytes (rituximab) require further evaluation.
Cystic Lung Disease
The HRCT pattern in SS-ILD is not specific. However, cystic lesions are reported in about 30 % of the patients with SS, sometimes associated with LIP [145] (Figs. 26.11 and 26.14). It has been suggested that cysts form as a consequence of bronchiolar obstruction due to follicular bronchiolitis although this is uncertain [145, 146]. The presence of pulmonary cysts was associated with LIP by Ichikawa et al. in 1994 but cysts are also observed in SS without LIP [147]. The cysts are single or multiple, variable in size (from 5 to over 10 cm), with a thin or indiscernible wall. Their distribution is random, but they are often situated within the parenchyma. They sometimes include calcifications. With time, their size can reduce, remain stable, or increase. In rare cases, the cysts can become giant and cause chronic terminal respiratory failure [148]. In this context, the presence of nodules should also prompt a search for pulmonary lymphoma [149]. There is no specific treatment for cystic lung disease associated with SS, but lung transplantation has been performed in some cases.
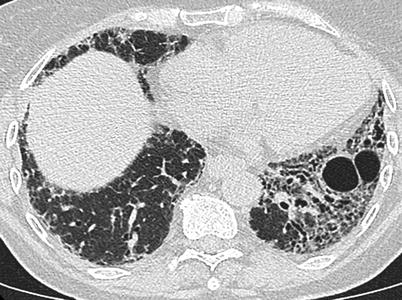
Fig. 26.14
A fibrotic pattern with lung cysts in a patient with primary Sjögren’s syndrome
Pulmonary Lymphoma
Patients with SS have an increased risk of developing a non Hodgkin lymphoma (relative risk: 6.5) [150, 151]. Sjögren’s associated lymphoma is usually a B-cell non Hodgkin’s lymphoma which arise primarily in the salivary glands, but also in mucosal sites including stomach and the lung. Pulmonary involvement occurs in 20 % of the patients with Sjögren’s associated lymphoma [152]. Radiographical presentation may vary: chronic alveolar opacities, reticular or reticulonodular opacities, diffuse nodular lesions, or pleural effusion with or without mediastinal disease [153–155]. On high-resolution CT, cysts are characteristic in patients with lymphocytic interstitial pneumonia, whereas consolidation, large nodules, and pleural effusions are characteristic in patients with malignant lymphoma (Fig. 26.15). These findings on high-resolution CT help differentiate LIP from malignant lymphoma [156]. Pulmonary lymphoma may be indolent and surgically removed, may be controlled with cytotoxic drugs such as chloraminophene or cyclophosphamide, and may evolve to an aggressive disease requiring a systemic polychemotherapy with monoclonal B-cell antibodies [153–155, 157]. The nature and existence of pseudolymphoma, a tumor-like aggregate of lymphoid cells that does not meet the criteria for malignancy, is debated.
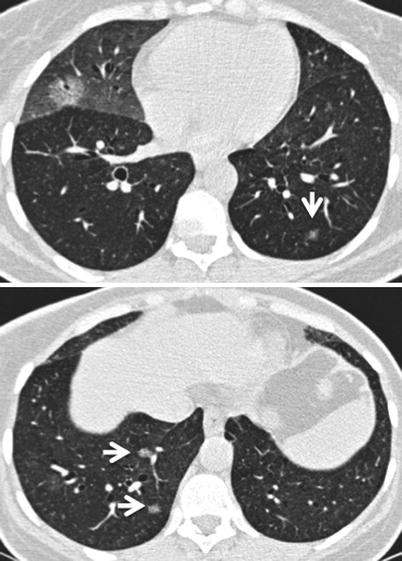
Fig. 26.15
Multifocal primary lung lymphoma in a patient with primary Sjögren’s syndrome. Small nodules are marked with arrows in upper and lower panel
Pulmonary Amyloidosis
Amyloid-associated cystic lung disease presenting as nodules, sometimes calcified, in conjunction with cystic lesions, has been detected in a limited number of patients [149, 158]. Pulmonary amyloid deposits are composed of amyloid light chains or AA protein, the most often localized in the lung, and exceptionally associated with systemic amyloidosis [159]. Mucosa-associated lymphoid tissue (MALT) lymphoma is associated in about 50 % of the patients [158]. PET scan performed in six patients did not reveal (18)F-2-deoxyglucose (FDG) uptake except in one nodule with borderline uptake [158].
Coexistence of Sjögren’s Syndrome and Pulmonary Sarcoidosis
The revised classification criteria for SS exclude patients with sarcoidosis [117]. However, cases of SS coexisting with sarcoidosis have been described and clinically well recognized [160] (Fig. 26.16). A recent review regrouped 59 observations from the literature reporting the coexistence of these two diseases in the same patient [161]. The frequency of sarcoidosis during primary SS has been estimated at 1 % [160, 161], which is far higher than the prevalence of sarcoidosis in the general population [162] and argues in favour of a pathophysiological connection between the two diseases, both characterized by considerable lymphocyte activation and a high prevalence of HLA-DR3. The diagnosis is made on the presence of antinuclear antibodies with anti-SSA or anti-SSB antibodies and non-granulomatous lymphocytic sialoadenitis in a patient with sarcoidosis [161]. The patients are the most often women (83 %). Mean age is 50. Both diseases are diagnosed at the same time in 60 % of cases. In 20 % of cases, the diagnosis of SS precedes that of sarcoidosis by 4 years on average. In 20 % of cases, the sarcoidosis is diagnosed on average 8 years before the SS [161]. Severe pulmonary hypertension with or without ILD [163] has been also reported.
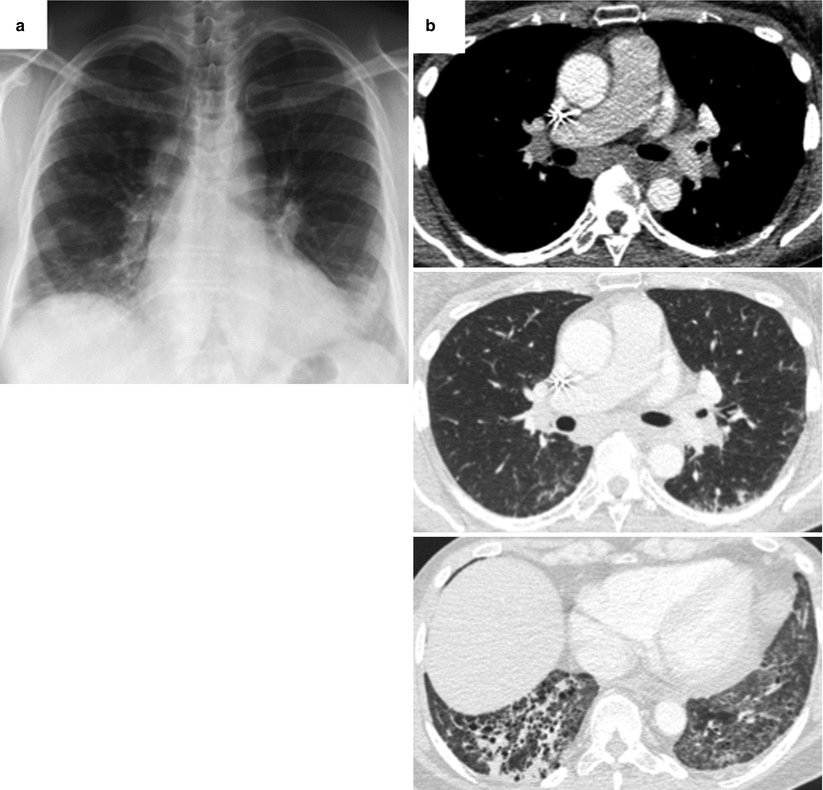
Fig. 26.16
Association of sarcoidosis and non specific interstitial pneumonia in a patient with primary Sjögren’s syndrome. Chest X ray (panel a) and CT scan (panel b) demonstrate hilar and mediastinal adenomegaly, and a combination of reticulations and ground glass opacities with a subpleural and lower zones predominance. Surgical sampling of mediastinal lymph nodes through mediastinoscopy evidenced extensive non necrotizing tuberculoid granulomas. Lung tissue was obtained through videothoracoscopy and showed a pattern of non specific interstitial pneumonia
Polymyositis and Dermatomyositis
Among the inflammatory myopathies, polymyositis and dermatomyositis are particularly associated with pulmonary involvement. While both polymyositis and dermatomyositis can share the same repertoire of specific antibodies and possess similar clinical patterns of muscle involvement, their divergent muscle histopathology suggests a different immunopathogenesis [164] (see Table 26.4 for classification criteria). Polymyositis appears to be a CD8+ predominant T-cell mediated assault on the myofiber, suggesting that the targeted antigen lies within the muscle surface. In contrast to polymyositis, dermatomyositis appears to be humorally mediated, with B-cells and CD4+ T cells located in a perivascular pattern leading to a perifascicular atrophy and fibrosis. Dermatomyositis and polymyositis are characterized by skeletal muscle inflammation and often involve the skin and lungs [113]. Skin findings in dermatomyositis include a violaceous erythematous rash over the interphalangeal joints, knuckles, elbows or knees (Gottron sign), eyelids (heliotrope rash), or nape of the neck and upper chest or upper back and shoulders [165]. Some patients have amyopathic dermatomyositis characterized by minimal to no muscle involvement yet severe ILD [166]. Pulmonary involvement may precede by many years, or occur simultaneously or follow the muscular manifestations of inflammatory myopathies [167, 168]. The variable and subtle presentations of these patients, requiring a high index of suspicion for the presence of ILD in the setting of myositis and for dermatomyositis or polymyositis in patients with seemingly idiopathic ILD or ARDS (see Table 26.6 for suggested investigations in a patient with ILD and suspected myositis). In addition to creatine kinase and electromyogram, detection of inflammatory lesions by f-18 fluorodeoxyglucose positron emission tomography [169] or MRI of proximal muscles [170] may aid in diagnosis and guide muscle biopsy.
Table 26.4




Diagnostic criteria for Dermatomyositis and Polymyositis [204]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
