Immunologic and Interstitial Diseases |
INTRODUCTION
Commonly, interstitial lung disease (ILD) presents with dyspnea on exertion, diffuse bilateral infiltrates on chest imaging, and restriction with diffusion impairment on physiologic testing. When tissue is obtained, the lung parenchyma may contain any combination of abnormalities, including inflammation, fibrosis, and granulomas. While many forms of ILD are extremely rare, there are some, such as idiopathic pulmonary fibrosis (IPF) and sarcoidosis that are seen commonly in general pulmonary practice. Masqueraders of ILD such as infection, pulmonary edema, and malignancy will be encountered in the assessment of an abnormal chest radiograph, and distinguishing these from true ILD is crucial.
ILD refers to a heterogeneous collection of more than one hundred distinct lung disorders that tend to be grouped together because they share clinical, radiographic, and pathologic features. These disorders are sometimes called diffuse parenchymal lung disease (DPLD) to make the point that the interstitium is not the only compartment of the lung affected. Entities such as organizing pneumonia or pulmonary alveolar proteinosis may cause an alveolar filling process. Respiratory bronchiolitis and chronic hypersensitivity pneumonitis may center on the airway, involving this compartment as well. Occasionally, purely airway-centered diseases like bronchiolitis obliterans may be initially identified as an ILD because of overlapping radiographic findings. A structured approach is necessary since treatments may vary considerably depending on the diagnosis.
Diagnosis is based upon a comprehensive history, a careful physical examination, as well as review of laboratory data, physiologic studies, radiography, and in some cases, pathologic tissue obtained from lung biopsy. Multidisciplinary review is an important part of the process and can have a significant impact on diagnostic and management decisions. For each patient, decisions regarding diagnostic approach and therapy must be individualized based upon the patient’s respiratory status, comorbid medical conditions, and personal approach to medical care.
DIAGNOSTIC APPROACH TO ILD
Several classification schemes for ILD have been proposed, which include histopathologic and clinical characteristics.1 The American Thoracic Society (ATS)/European Respiratory Society (ERS) consensus panel classification system was published in 2001 and has recently been revised.2 One approach is given in Figure 54-1.
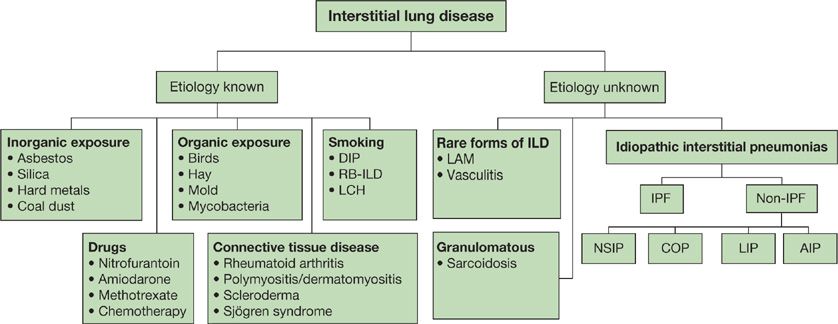
Figure 54-1 An overview of ILD classification. DIP, desquamative interstitial pneumonia; RB-ILD, respiratory bronchiolitis interstitial lung disease; LCH, Langerhans cell histiocytosis; LAM, lymphangioleiomyomatosis; IPF, idiopathic pulmonary fibrosis; NSIP, nonspecific interstitial pneumonia; COP, cryptogenic organizing pneumonia; LIP, lymphocytic interstitial pneumonia; AIP, acute interstitial pneumonia. (Data from ATS/ERS classification, 2001.)
It is often helpful in the case of any individual patient to incorporate a combination of historical, clinical, radiographic, and pathologic features. Distinguishing those patients who have a known cause for their ILD (e.g., connective tissue disease, occupational or environmental exposure, or drug toxicity) from those who do not (e.g., IPF, sarcoidosis) is an important first step. Clinical features, such as acuity of onset, may help to classify disease as well. For example, the subacute time course of a patient with organizing pneumonia will be quite different than the insidious onset of dyspnea in IPF. The radiographic pattern of IPF will be characterized by bibasilar traction bronchiectasis and honeycombing, while sarcoidosis may demonstrate upper lobe-predominant nodular opacities with retraction and volume loss. Histopathologic characterization may range from acute inflammation to granulomatous involvement, or fixed fibrosis and collagen deposition.
It is important to note that the names used to describe the clinical entities themselves must be distinguished from the radiographic and pathological terms used to describe the findings. For example, IPF is the clinical entity associated with a usual interstitial pneumonia (UIP) pattern on chest CT and on pathology. However, the finding of a UIP pattern alone does not ensure a diagnosis of IPF, as this may also be seen in other entities, such as the connective tissue diseases.
CLINICAL HISTORY
The typical presentation of ILD is nonspecific and may include vague pulmonary complaints, such as dyspnea on exertion or cough, and an abnormal radiograph. In some cases the time course of the disease may suggest certain forms of ILD (Table 54-1). The acute forms of ILD must be distinguished from respiratory infections and pulmonary edema due to congestive heart failure.
It is important to consider key features of the patient. For example, the differential diagnosis for dyspnea and diffuse infiltrates on chest radiograph will vary markedly between immunocompetent patients and those who have undergone organ or bone marrow transplantation, are neutropenic from recent chemotherapy, or who have advanced HIV disease. In the immunocompetent host, it is often after a lack of response to antibiotics or diuretics that ILD is suspected. Other clinical features may affect the presentation as well. In elderly or disabled patients, musculoskeletal issues may limit mobility and dyspnea may be a late symptom. In a patient with other cardiopulmonary disease, relatively minor ILD may cause an earlier onset of disabling dyspnea and the discovery of ILD for this reason. It is helpful to attempt an objective quantification of the degree of dyspnea, such as the distance the patient walks before becoming breathless or the number of steps climbed before a rest must be taken.
Often a persistent cough after a respiratory infection leads to the initial chest radiograph. While other respiratory symptoms are less common, they may help focus the differential diagnosis. For example, a history of wheezing may suggest an airway-centered process such as hypersensitivity pneumonitis, eosinophilic pneumonia, or sarcoidosis.3 Substernal chest pain is commonly described by patients with sarcoidosis.4 Pleuritic chest pain may herald serositis in a patient with connective tissue disease, or pneumothorax in cystic lung diseases such as lymphangioleiomyomatosis (LAM) and Langerhans cell histiocytosis (LCH).5–7 Hemoptysis may suggest diffuse alveolar hemorrhage, though it is not present in most cases.8
SYSTEMIC SYMPTOMS
Connective tissue disease is a frequent cause of ILD, and patients may come with a pre-existing diagnosis. However, most will not and it is incumbent on the physician to search carefully for suggestions of underlying autoimmune disease, as the pulmonologist may be the first to make such a diagnosis, prompted by the onset of ILD. In some cases, nonspecific systemic symptoms such as night sweats, fever, fatigue, or weight loss suggest an underlying inflammatory condition. In others, one can arrive at a specific diagnosis by merely performing a thorough review of systems. For example, careful questioning regarding dermatologic symptoms may lead to the discovery of dermatomyositis as demonstrated by a heliotrope rash, Gottron’s papules, or “mechanic’s hands,” all of which may be relatively specific features that do not bother the patient. Patients with underlying systemic sclerosis (scleroderma) may give a history of skin tightness and thickening, telangiectasias, Raynaud’s phenomenon, or digital pitting. Papular eruptions, lupus pernio, and erythema nodosum may be seen in sarcoidosis.9 Patients with systemic lupus erythematosus (SLE) may describe malar rash, photosensitivity skin reaction, or hair loss.
Gastrointestinal symptoms may be indicative of underlying esophageal motility problems related to connective tissue disease such as systemic sclerosis and polymyositis, or may be the root cause of the ILD itself. In particular, symptoms suggestive of acid reflux (chest burning or pressure, cough after meals, regurgitation of food) should be sought. Chronic, intermittent aspiration can lead to progressive fibrotic lung disease from recurrent lung injury. Patient descriptions of coughing or choking while eating or noting food “going down the wrong pipe” are suggestive of frank aspiration and could lead to a discovery of progressive neuromuscular diseases such as amyotrophic lateral sclerosis (ALS), cerebrovascular accidents, or other causes of oropharyngeal and laryngeal dysfunction.10 Other gastrointestinal complaints, such as bloating and diarrhea, may suggest inflammatory bowel disease or bacterial overgrowth due to bowel dysmotility in systemic sclerosis.
Musculoskeletal complaints can be helpful in identifying underlying connective tissue disease as well. In particular, arthralgias, morning stiffness, joint swelling and erythema, and deformities may be evidence of an underlying inflammatory disorder such as rheumatoid arthritis, Sjögren syndrome, or mixed connective tissue disorder. Swollen fingers (“sausage digits”) may be observed in systemic sclerosis and polymyositis. Raynaud’s phenomenon manifests as either blue/purple discoloration or whiteness of the digits (fingers or toes) in the cold. In some cases this may be quite profound and can even be associated with digital ulcerations and, rarely, digital gangrene. This finding is most suggestive of underlying scleroderma, mixed connective tissue disease, SLE, and antisynthetase syndrome.11
Ophthalmologic symptoms may direct the clinician toward particular clinical entities. Inquiries regarding dry eyes or the use of eye drops may uncover sicca syndrome, as seen in Sjögren syndrome and overlap connective tissue diseases. Patients with a history of uveitis may have underlying SLE or sarcoidosis. Neurologic symptoms may suggest vasculitis or sarcoidosis.
A detailed review of systems may uncover sequelae of longstanding ILD. Increasing edema, syncopal events, or exertional chest discomfort may indicate severe pulmonary hypertension and cor pulmonale in the patient with advanced fibrotic lung disease and hypoxemia. Alternatively, in a patient with systemic sclerosis, these findings might indicate a second primary problem such as pulmonary arterial hypertension. The presence of palpitations or syncope in a patient with sarcoidosis can lead to a diagnosis of cardiac sarcoidosis.
The review of systems will sometimes also lead to a related secondary diagnosis. For example, more typical causes of exertional chest discomfort, such as cardiac ischemia, may be truly present and may be exacerbated by exertional desaturation. This is particularly important to remember, as patients with underlying immune-related diseases appear to be at increased risk for coronary artery disease.12 In addition, pleuritic chest pain, leg swelling, and increasing dyspnea should prompt consideration of acute pulmonary embolism, as patients with ILD are at increased risk for this complication.13,14
PAST MEDICAL HISTORY
As mentioned, a prior diagnosis of connective tissue disease (systemic sclerosis, rheumatoid arthritis, SLE) is extremely pertinent for patients with ILD. In the case of HIV disease, specific forms of lung disease such as lymphocytic interstitial pneumonia (LIP) are more commonly observed. A prior history of acute or chronic kidney disease might suggest underlying vasculitis, pulmonary–renal syndromes, or connective tissue disease. A history of liver disease could suggest sarcoidosis, primary biliary cirrhosis, or underlying genetic abnormalities resulting in short telomere length.15–17 A history of facial nerve paralysis (Bell’s palsy) might represent unrecognized sarcoidosis.18
OCCUPATIONAL HISTORY
The occupational history should be thorough, including a review of all jobs held in the past. In particular, attention should be paid to any occupation in which a history of exposure to organic or inorganic products was present (Table 54-2). Specific inquiries may be made regarding a history of construction work, including demolition, plumbing, and electrical work. The patient should be questioned about employment in factories and manufacturing plants, the electronics industry, metal working, stone cutting, and mining. The clinician should specifically inquire about exposure to asbestos, silica, hard metals, and beryllium, as workers often know when these have been present in their work environment.19,20 Inquiries may additionally be made regarding the jobs held by the household contacts of patients. For example, spouses may have been subjected to significant levels of dust inhalation via clothing worn by the worker.21 In cases where a relevant exposure has occurred, details should be obtained regarding the worker’s specific duties, the typical proximity to the exposure of interest and any use of respiratory protection. In some cases, related respiratory illnesses in coworkers can raise suspicion. There may be a long latency period between the exposure and the onset of symptoms and radiographic changes, so the occupational history must explore jobs held many years in the past. A history of farm work or other agricultural employment should raise suspicion for an environmental cause of ILD. Exposure to moldy hay, bird feathers and droppings, and a variety of organic products can lead to chronic hypersensitivity pneumonitis.

ENVIRONMENTAL HISTORY
Organic exposures are also frequently encountered in household and office settings. For example, humidification systems may be contaminated with mold.22 Hot tubs and other aerosolized water sources have led to lung disease related to the growth of Mycobacterium avium.23 The exposure history should include a thorough inquiry regarding the home heating and humidification system, a history of water damage, or visible evidence of significant mold growth on walls. An assessment of the timing of any water damage relative to the onset of the lung disease can be helpful. Domestic birds are a common source of feathers and dropping antigen. Bird owners often have multiple birds and have owned birds for many years. Frequently, these pets are considered to be cherished members of the family, residing in bedrooms and main living spaces. A history of hobbies and materials used should also be obtained.
Cigarette smoke is one of the most common environmental exposures and is strongly linked with several forms of ILD, including desquamative interstitial pneumonitis (DIP), respiratory bronchiolitis-ILD, and LCH. Cigarette smoking has been identified as a risk factor for IPF.
MEDICATION HISTORY
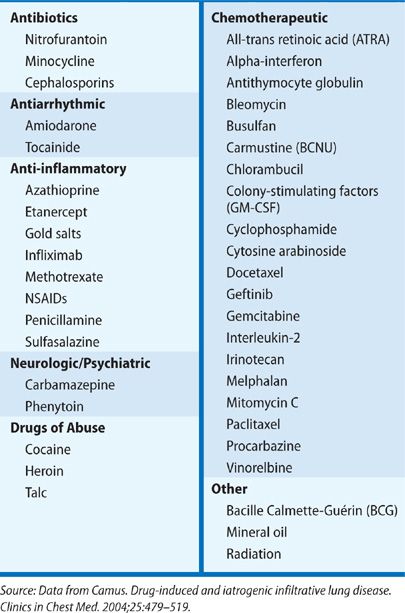
Numerous drugs have been implicated in the development of ILD, ranging from acute pneumonitis to chronic fibrotic lung disease (Table 54-3). The use of several commonly prescribed drugs should be sought. Nitrofurantoin, used for suppression of recurrent urinary tract infections, may lead to severe ILD.24 Amiodarone, used for management of atrial and ventricular arrhythmias, is known to lead to lung toxicity.25 Nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly used and may lead to eosinophilic and other inflammatory lung disease.26 While less frequently used in the general population, a history of recent chemotherapy should trigger suspicion for drug-related lung disease.27 Similarly, a history of immune-modulating drug use, such as in the treatment of rheumatoid arthritis or Crohn’s disease is suspicious.24,25,28 So many drugs have been reported in association with ILD that it is recommended to review the patient’s entire medication history thoroughly with this in mind. A helpful online source of information is: http://www.pneumotox.com. This independent resource is compiled and regularly updated by members of the University Hospital in Dijon, France, and the GEPPI (Groupe d’Etudes de la Pathologie Pulmonaire Iatrogène). It lists all reports of suspected drug-induced lung disease, categorizes the radiographic patterns observed, and gives an assessment of the quality of the compiled information.
FAMILY HISTORY
Most forms of ILD are not heritable, though several do have a genetic component. When heritable disease is suspected, it is important to consider both systemic disorders as well as those that primarily affect the lung. In particular, by considering a patient’s concomitant medical history or the history of family members, specific diagnoses may be uncovered. Several inborn errors of metabolism (Gaucher disease and Niemann-Pick, for example) are inherited in an autosomal recessive fashion. Other rare diseases such as Hermansky–Pudlak syndrome, Burt–Hogg–Dubé syndrome, and neurofibromatosis type I are autosomal dominant disorders.29 The cystic lung disease, LAM may be associated with mutations in the tuberous sclerosis complex (TSC) genes, in which systemic hamartomas are present, including a high incidence of renal angiomyolipomas.29
The telomere shortening syndromes that lead to dyskeratosis congenita may lead to a phenomenon known as genetic anticipation, in which telomere lengths are progressively shortened in subsequent generations. This leads to the earlier onset of more severe disease in each subsequent generation. In the case of IPF, mutations in the telomerase genes have been demonstrated among 8% to 15% of familial cases and among 1% to 3% of patients with sporadic disease.17 Several other genes have been linked with the onset of IPF, including those involved in the regulation of surfactant protein C and MUC5B.30,31 There may be familial predisposition to sarcoidosis, but inheritance is more complex, involving an interaction of genetic and environmental factors.32
PHYSICAL EXAMINATION
Most patients with pulmonary fibrosis have lung findings characterized by fine, inspiratory, basilar “Velcro” crackles and many will have digital clubbing. In contrast, patients with nonfibrotic lung disease may have clear lung fields on auscultation. The location of the abnormal breath sounds may be suggestive of the underlying diagnosis, such as the upper lobe findings in silicosis and sarcoidosis in contrast to the lower lobe abnormalities in IPF. Additional breath sounds such as wheezing and inspiratory squeaks can indicate airway disease, which may focus the examiner’s differential diagnosis on airway-centered diseases such as bronchiolitis, sarcoidosis, and hypersensitivity pneumonitis. These additional sounds should not be present in a patient with IPF.
Other cardiopulmonary manifestations of disease should also be sought. Signs of pulmonary hypertension and right heart failure include an increased P2 component, a right ventricular heave, elevated jugular venous pressure, and lower extremity edema.
Dermatologic and musculoskeletal signs of connective tissue disease, including skin rashes, sclerodactyly, skin thickening, digital ulceration, “mechanic’s hands,” synovitis, joint deformities, Raynaud’s phenomenon, and telangiectasias, may be extremely helpful in narrowing the differential diagnosis. Less common findings such as cutaneous neurofibromas or café-au-lait spots in neurofibromatosis, albinism in Hermansky–Pudlak syndrome, and facial angiofibromas, periungual fibromas, and Shagreen patch in TSC may all be key features only recognized through a careful and focused search.
CHEST IMAGING
Findings on chest radiography and high-resoluaion computed tomography are discussed below.
 CHEST RADIOGRAPH
CHEST RADIOGRAPH
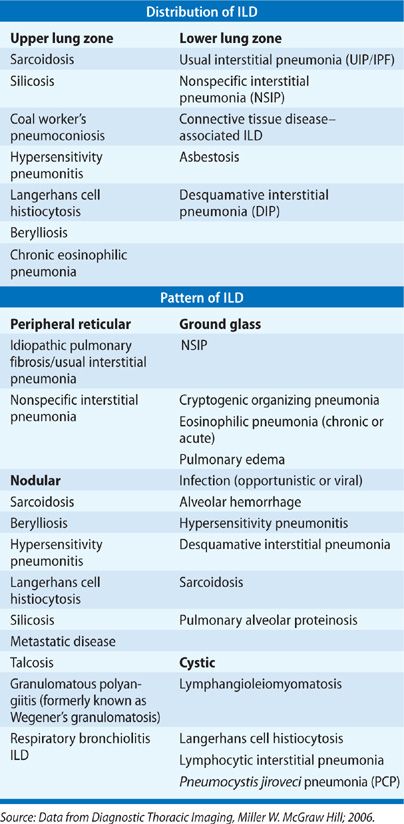
An abnormal chest radiograph is often the first indication of underlying ILD. The pattern and distribution of abnormalities often help in formulating a differential diagnosis (Table 54-4). For example, sarcoidosis, silicosis, and LCH are among the diseases with an upper lobe predominance, while IPF, connective tissue disease–associated ILD, and asbestosis are all lower lobe predominant. Peripheral alveolar opacities are typical findings in organizing pneumonia and chronic eosinophilic pneumonia. The chest radiograph is also helpful for assessing lung volumes. In particular, fibrotic lung disease such as IPF leads to small lung fields whereas lung volumes are maintained and may even demonstrate hyperinflation in diseases such as LAM and LCH. In many cases, the presence of lymphadenopathy, with hilar fullness observed on chest radiograph, is the feature that leads to an accurate diagnosis, such as in sarcoidosis.
 HIGH-RESOLUTION CHEST COMPUTED TOMOGRAPHY
HIGH-RESOLUTION CHEST COMPUTED TOMOGRAPHY
High-resolution computed tomography (HRCT) of the chest is significantly more sensitive than chest radiograph for abnormalities in ILD. Characteristic patterns observed on HRCT can be quite specific in some cases. For example, the characteristic radiographic features of IPF are collectively known as the “UIP pattern,” since these features have been demonstrated to confidently predict the presence of pathologic UIP when surgical biopsy is obtained (Table 54-5).33,34 This pattern consists of peripheral, subpleural, basilar-predominant reticular opacities in combination with basilar honeycombing and without features, such as ground-glass opacities, cysts or nodules, to suggest another form of ILD.35 When these features are accurately recognized, patients can be spared a surgical biopsy for diagnosis (Fig. 54-2).
Source: Data from Schmidt. Respirology. 2009;14(7):934–939.
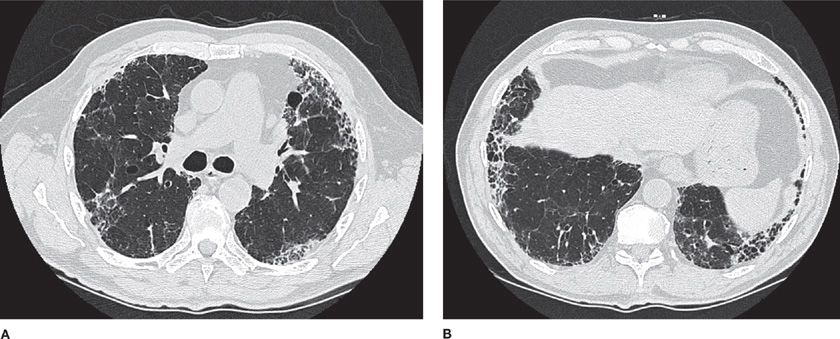
Figure 54-2 An 82-year-old man with progressive dyspnea and a usual interstitial pneumonia (UIP) radiographic pattern. High-resolution (1.25-mm thick sections) CT images at the level of the midthorax (A) and lower thorax (B) show peripheral reticular markings with architectural distortion and small subpleural cysts/honeycombing. Ground-glass opacities and other features atypical for IPF are absent. The patient had no exposures and no clinical evidence of connective tissue disease. The final diagnosis was idiopathic pulmonary fibrosis (IPF). No lung biopsy was performed, as a diagnosis was made based upon CT criteria alone.
Several radiographic patterns have been identified that help to focus the differential diagnosis (Table 54-4). In particular, a peripheral reticular pattern, that is basilar predominant may be seen in IPF, NSIP, and connective tissue disease–associated ILD (Fig. 54-3). Nodular infiltrates may suggest sarcoidosis, hypersensitivity pneumonitis, and LCH, though the nature of the nodules and their location may make one or another diagnosis more likely (Fig. 54-4). The finding of diffuse cystic abnormalities leads to a specific differential diagnosis, including LAM, LCH, and LIP. The term “ground glass” refers to areas of lung tissues with increased attenuation, but not enough to obscure or distort lung architecture, blood vessels, and lymphatics. Alveolar opacities are a related finding and reflect more dense attenuation of lung tissue, sometimes containing air bronchograms. Many forms of ILD are characterized by ground-glass and alveolar opacities. When observed in characteristic distributions, such as the peripheral, patchy alveolar opacities in cryptogenic organizing pneumonia (COP) and chronic eosinophilic pneumonia, they are quite suggestive of an underlying pathologic pattern (Fig. 54-5).36 However, the radiographic appearance in such cases is nonspecific and other testing, including lung biopsy, may be required for diagnosis.
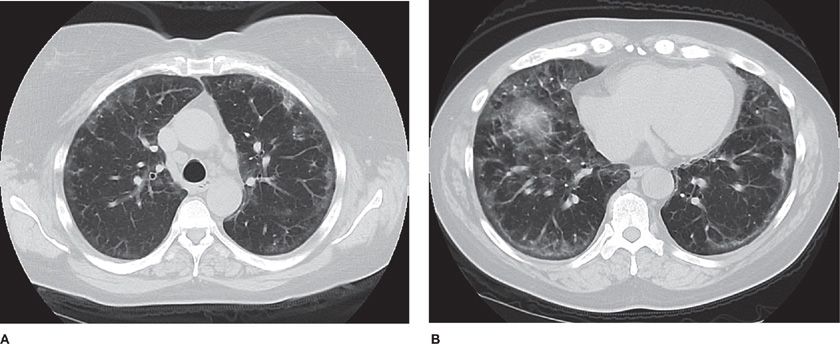
Figure 54-3 A 55-year-old woman with progressive dyspnea on exertion. She had finger swelling and Raynaud phenomenon, with an antinuclear antibody titer of 1:2560 (nucleolar). Axial CT image through the midthorax (A) and lower thorax (B) show ground-glass opacities in a peripheral distribution, reticular markings, and mild architectural distortion. Subpleural sparing is evident. These findings are all compatible with a nonspecific interstitial pneumonia (NSIP) pattern. The final diagnosis was scleroderma lung disease.
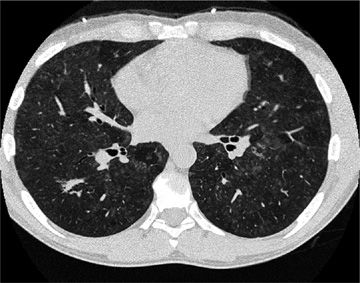
Figure 54-4 A 47-year-old man who enjoyed soaking in the hot tub after exercising at the gym developed dyspnea and hypoxemia. He improved clinically with removal from the presumed source of exposure. High-resolution CT images at the level of the midthorax demonstrate diffuse centrilobular ground-glass nodules as well as areas of lobular lucency, consistent with air trapping. These findings are consistent with subacute hypersensitivity pneumonitis.
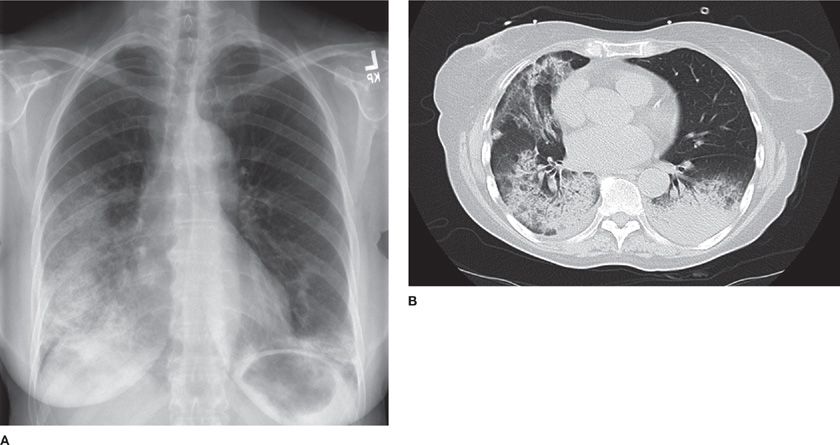
Figure 54-5 A 58-year-old woman with organizing pneumonia (OP) secondary to radiation for breast cancer. Frontal chest radiograph (A) demonstrates ground-glass and alveolar opacities. High-resolution CT images at the level of the midthorax (B) demonstrate both ground-glass opacities and areas of consolidation. She had complete response to corticosteroid therapy.
LABORATORY TESTING
Routine laboratory testing, while often normal, may lead to the diagnosis of a previously unrecognized systemic disease, or may be suggestive of the underlying lung disease. Its importance may also lie in the discovery of associated conditions, such as elevated liver enzymes or hypercalcemia in sarcoidosis, or renal insufficiency in pulmonary renal syndromes and other connective tissue disease with renal involvement. Other examples include the presence of peripheral eosinophilia in chronic eosinophilic pneumonia, Churg–Strauss syndrome, drug reaction, and other forms of vasculitis. Anemia may worsen symptoms of dyspnea in the setting of ILD and may reflect an underlying hemolytic condition, or chronic gastrointestinal blood loss in inflammatory bowel disease. In addition to eliciting causes for the ILD, recognition of underlying chronic organ dysfunction will also help determine the ability of the patient to tolerate treatment.
More advanced testing is often necessary, including serologic testing for underlying connective tissue disease (Table 54-6). Many of the tests listed here are employed by ILD centers in the evaluation of patients newly presenting with diffuse lung disease, though no clear standard exists. Importantly, ILD can be the sole manifestation or the presenting feature of a connective tissue disease, making a careful evaluation for previously unrecognized autoimmune disease essential.37



