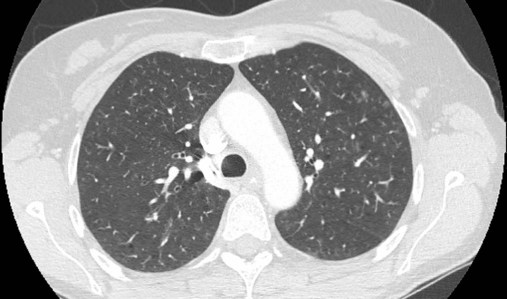
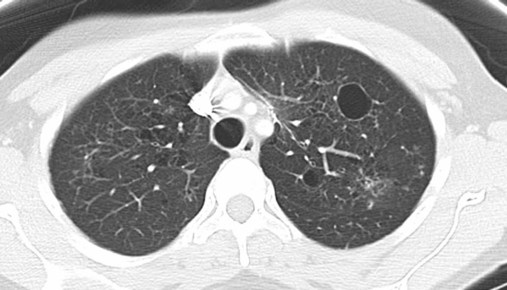
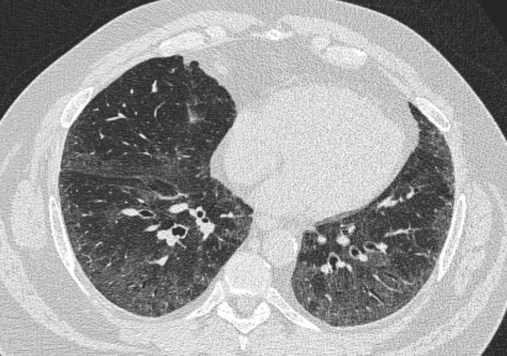
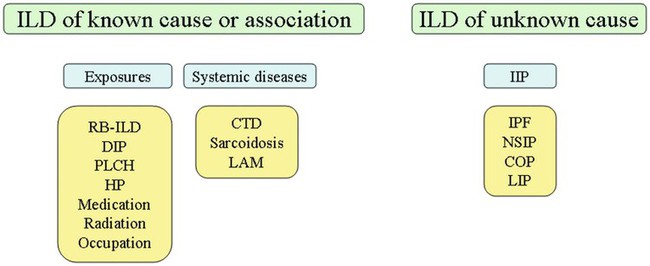
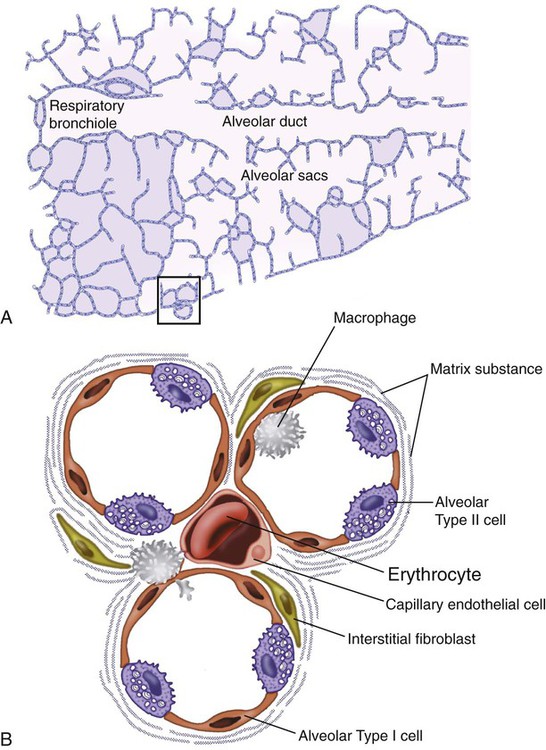
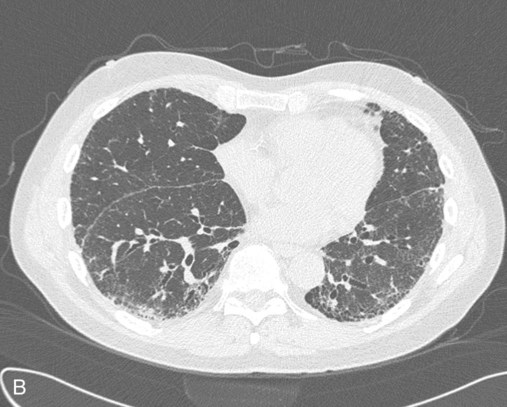
After reading this chapter you will be able to: Interstitial lung disease (ILD) comprises a broad category of lung diseases rather than a specific disease entity.1,2 This category includes various illnesses affecting the lung parenchyma with many different causes, treatments, and prognoses. These disorders are grouped together because of similarities in their clinical presentations, appearance on plain chest radiography, and physiologic features. With over 100 separate disorders, it is essential to group ILDs based on etiology, disease associations, or pathology. An organizational scheme is presented in Figure 24-1. When evaluating patients with an ILD, one first must consider diseases with known causes or associations, such as diseases related to specific exposures, diseases associated with systemic conditions, and diseases with a known genetic basis. Most patients do not have an ILD with a known cause, and their disorder is classified by pathologic pattern. These groups are divided into specific disease entities. Using this organizational scheme to guide a careful and complete history, one is able to understand the disease processes and work efficiently toward an accurate diagnosis. As the name ILD implies, the histologic abnormalities that characterize ILD involve the pulmonary interstitium to a greater extent than the alveolar spaces or airways, although exceptions exist. Figure 24-2 illustrates the components of the normal pulmonary parenchyma. The interstitium is the area between the capillaries and the alveolar space. As Figure 24-2 shows, in the normal state, this space allows close apposition of gas and capillaries with minimal connective tissue matrix, fibroblasts, and inflammatory cells such as macrophages. The interstitium supports the delicate relationship between the alveoli and capillaries allowing for efficient gas exchange. When responding to any injury—whether from a specific exposure (e.g., asbestos, nitrofurantoin, or moldy hay), an autoimmune-mediated inflammation from a systemic connective tissue disease (e.g., rheumatoid arthritis), or unknown injury (e.g., idiopathic pulmonary fibrosis [IPF])—the lung must respond to the damage and repair itself. If the exposure or injury persists or if the injury repair process is imperfect, the lung may be permanently damaged with increased interstitial tissue replacing the normal capillaries, alveoli, and healthy interstitium. Many ILDs have similar clinical features and are not easily distinguished based on history or examination. Symptoms are generally limited to the respiratory tract. However, extrapulmonary symptoms should not be ignored because they may point to an ILD associated with a systemic diagnosis. Exertional breathlessness (dyspnea) and a nonproductive cough are the most common reasons patients seek medical attention. Other respiratory symptoms, such as sputum production, hemoptysis, pneumothorax, or wheezing, can occur and be suggestive of specific diseases (Table 24-1). If the patient also has prominent extrapulmonary symptoms, such as myalgia, arthralgia, sclerodactyly, gastroesophageal reflux, or Raynaud phenomenon, ILD resulting from underlying connective tissue disease may be present (Table 24-2). TABLE 24-1 Unusual Pulmonary Findings and Likely Diagnosis TABLE 24-2 Extrapulmonary Findings and Likely Diagnosis ILDs manifest as abnormal lung parenchyma that cast abnormal radiographic shadows. For most ILDs, the chest radiograph reveals reduced lung volumes with bilateral reticular or reticulonodular opacities. However, the chest radiograph has limited value because the three-dimensional abnormalities are summed into a two-dimensional image with loss of spatial information. High-resolution cross-sectional imaging via computed tomography (CT) provides detailed images representing pulmonary pathology.3 High-resolution CT images allow noninvasive evaluation of ILDs and are a key element in making a confident diagnosis and managing ILD.4 Plain chest radiographs and high-resolution CT images of usual interstitial pneumonitis (UIP) show the prototypic fibrotic injury pattern. The chest radiograph (Figure 24-3, A) and high-resolution CT image (Figure 24-3, B) in UIP typically reveal a bilateral, patchy, peripheral (subpleural), and basilar predominant disease with reticulonodular infiltrates, often with honeycomb, cystic change. The lung architecture is distorted in patients with moderate or severe disease burden, with reduced lung volume and traction bronchiectasis, especially at the lung bases. Reticulogranular (ground-glass) abnormalities, increased attenuation of the lung tissue without distortion of the underlying blood vessels or bronchi, are absent or minimal in IPF. Pleural disease, air trapping, micronodules, and significant lymphadenopathy are not seen, although two-thirds of patients with IPF can have mild mediastinal adenopathy.5 Similar to the radiographic findings, there can be considerable variability among the specific diseases in the physiologic abnormalities seen. However, a restrictive physiologic impairment is the common finding.6 Both forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) are diminished, and the FEV1/FVC ratio is preserved or even supranormal. Lung volumes are reduced, as is the diffusing capacity of the lung for carbon monoxide (DLCO). This reduction in diffusing capacity reflects a pathologic disturbance of the alveolus-capillary interface. Although not commonly pursued, the compliance characteristics of the lungs can be evaluated with an esophageal balloon to measure intrathoracic pressure at various lung volumes. In almost all ILDs, the lungs have reduced compliance and require supranormal transpleural pressures to ventilate (Figure 24-4). This lack of compliance results in small lung volumes and increased work of breathing. Less frequently, a pattern of physiologic obstruction may be seen. This obstruction can be the result of the primary disease process (e.g., LAM, PLCH, or sarcoidosis in some patients) or concomitant emphysema or asthma.7 If ILD develops in a patient with significant emphysema, the opposing physiologic effects of the two diseases may result in deceptively normal spirometry and lung volume measurements and apparently normally compliant lungs. However, because both emphysema and ILD result in impaired gas exchange, DLCO is significantly decreased. Although the association of first-hand tobacco smoke and obstructive lung disease is common and well known, tobacco smoke is also an avoidable cause of ILD. Although the association is rarer than with obstructive lung disease, first-hand tobacco smoke inhalation can lead to three types of ILD in susceptible individuals: RB-ILD, DIP, and PLCH. The first two disorders are related. Each disease consists of increased numbers of polyclonal activated macrophages. The diseases differ by the location of these overly abundant cells. In RB-ILD, macrophages accumulate in the respiratory bronchioles leading to bronchiolar remodeling and fibrosis of adjacent alveoli. As expected for a disease with combined airway and alveolar injury, pulmonary function testing reveals mixed restriction and obstruction with frequent air trapping. High-resolution CT images show this mixed pathologic location with indistinct centrilobular nodules (Figure 24-5). In DIP, the increased macrophages fill the alveoli, manifesting as restrictive impairment on pulmonary function testing and diffuse ground-glass attenuation on high-resolution CT imaging (Figure 24-6). Pulmonary Langerhans cell histiocytosis (PLCH) is the third interstitial manifestation of tobacco smoke. Increased numbers of polyclonal macrophages play a prominent role. However, in PLCH, they are accompanied by fibroblasts and eosinophils in nodules concentrated around small airways. These nodules are stellate and destroy adjacent lung tissue, leading to the classic high-resolution CT image of stellate nodules associated with cysts as seen in Figure 24-7. Although adult smoking-associated PLCH is pathologically similar to childhood Langerhans cell histiocytosis, the adult form does not involve bone and has not proven to respond to chemotherapy as the childhood form does. The relationship of these two disorders has yet to be defined. In each of these three diseases, the primary treatment is complete tobacco abstinence. With abstinence, most patients either minimally improve or remain stable,8 but a few progressively worsen and may require lung transplantation. Active treatment with prednisone or other immunosuppressive medications is discouraged because few, if any, patients improve.9 Many drugs have been associated with pulmonary complications of various types, including interstitial inflammation and fibrosis, bronchospasm, pulmonary edema, and pleural effusions. Drugs from many different therapeutic classes can cause ILD, most commonly chemotherapeutic agents, antibiotics, antiarrhythmic drugs, and immunosuppressive agents (Box 24-1). There are no distinct physiologic, radiographic, or pathologic patterns of drug-induced ILD, and the diagnosis is usually made when a patient is exposed to a medication known to result in lung disease, the timing of the exposure is appropriate for the development of the disease, and other causes of ILD have been eliminated. Treatment is avoidance of further exposure and systemic corticosteroids in markedly impaired or declining patients. In addition to future drug exposure, bleomycin injury is accentuated by increased fractional inspired oxygen (FiO2), even months after last drug exposure, and supplemental oxygen (O2) should be used only if absolutely necessary in these patients.10
Interstitial Lung Disease
 Organize and distinguish between the entities grouped as interstitial lung diseases (ILDs).
Organize and distinguish between the entities grouped as interstitial lung diseases (ILDs).
 Interpret symptoms, examination signs, and pulmonary function testing in ILD.
Interpret symptoms, examination signs, and pulmonary function testing in ILD.
 List emerging pathophysiologic characteristics that are associated with selected ILDs.
List emerging pathophysiologic characteristics that are associated with selected ILDs.
 Describe how to manage ILD in general and how some specific ILDs can be treated.
Describe how to manage ILD in general and how some specific ILDs can be treated.
Characteristics of Interstitial Lung Disease
Clinical Signs and Symptoms of Interstitial Lung Disease
Pulmonary Findings
Disease
Frequency
Hemoptysis
LAM
Rare
Chyloptysis
LAM
Rare
Pneumothorax
LAM, BHD
Common
Wheeze
Sarcoidosis, LAM
Common
Chylous pleural effusion
LAM
Common
Exudative pleural effusion
RA
Common
Extrapulmonary Findings
Disease
Frequency
Raynaud phenomenon
All CTD
Common
Arthralgia
All CTD
Common
Myalgia
Polymyositis
Common
Large muscle weakness
Polymyositis
Common
Sclerodactyly
Scleroderma
Common
Rheumatoid skin nodules
RA
Rare
Fingertip fissures
Antisynthetase syndrome
Common
Dorsal hand rash
Dermatomyositis
Common
Facial rash
Dermatomyositis
Common
Exudative pleural effusion
RA
Common
Shawl distribution skin nodules
TSC
Common
“Pencil eraser” facial skin nodules
BHD
Common
Central nervous system benign cortical tuber
TSC
Common
Abdominal angiomyolipoma
LAM
Common
Renal cancer
BHD
Common
Cardiomyopathy
Sarcoidosis, polymyositis
Rare
Cardiac conduction block
Sarcoidosis
Rare
Violaceous facial skin nodules
Sarcoidosis
Common
Subcutaneous nodules
Sarcoidosis
Common
Cranial neuropathy
Sarcoidosis
Rare
Small fiber neuropathy
Sarcoidosis
Rare
Radiographic Features
Physiologic Features
Selected Specific Types of Interstitial Lung Disease and Therapies
Exposure-Related Interstitial Lung Disease
Tobacco-Associated Interstitial Lung Disease
Drug-Related and Radiation-Related Interstitial Lung Disease
Interstitial Lung Disease
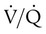 mismatching, shunt, and decreased diffusion across the abnormal interstitium. Work of breathing is markedly increased because of decreased lung compliance. Together, these physiologic impairments lead to the exercise intolerance seen in all of the ILDs. If the initiating injury or abnormal repair from injury is not halted, progressive tissue damage leading to worsening physiologic impairment and death can occur.
mismatching, shunt, and decreased diffusion across the abnormal interstitium. Work of breathing is markedly increased because of decreased lung compliance. Together, these physiologic impairments lead to the exercise intolerance seen in all of the ILDs. If the initiating injury or abnormal repair from injury is not halted, progressive tissue damage leading to worsening physiologic impairment and death can occur.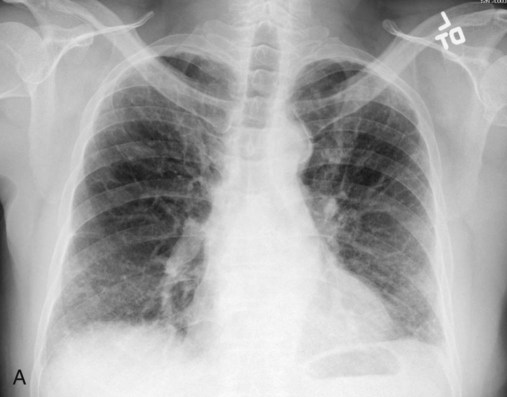
 Rule of Thumb
Rule of Thumb Rule of Thumb
Rule of Thumb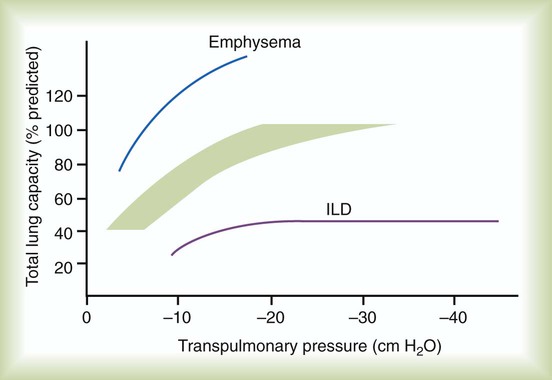
 Rule of Thumb
Rule of Thumb