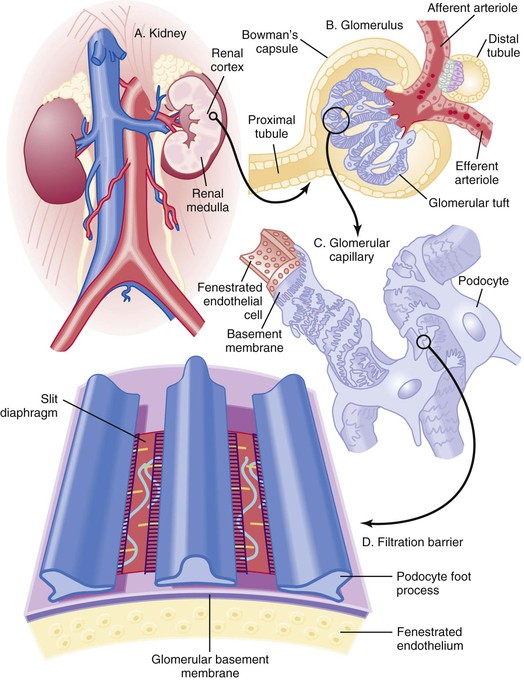
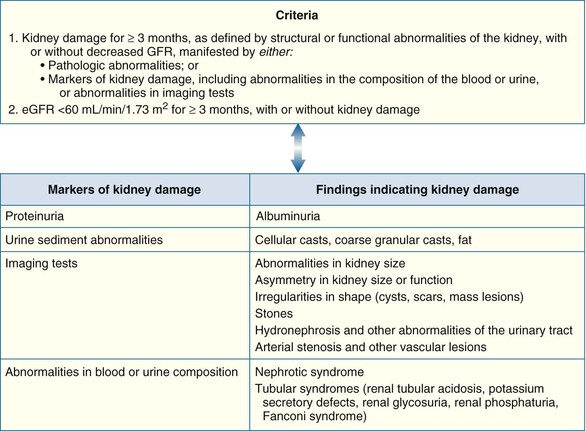
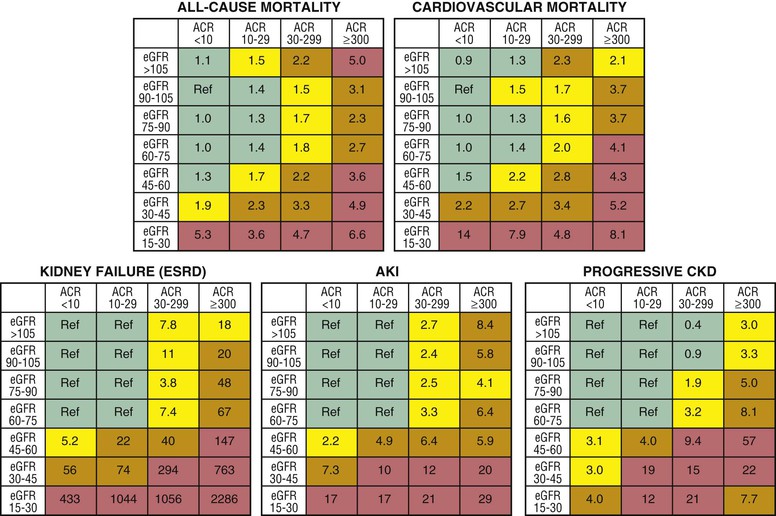
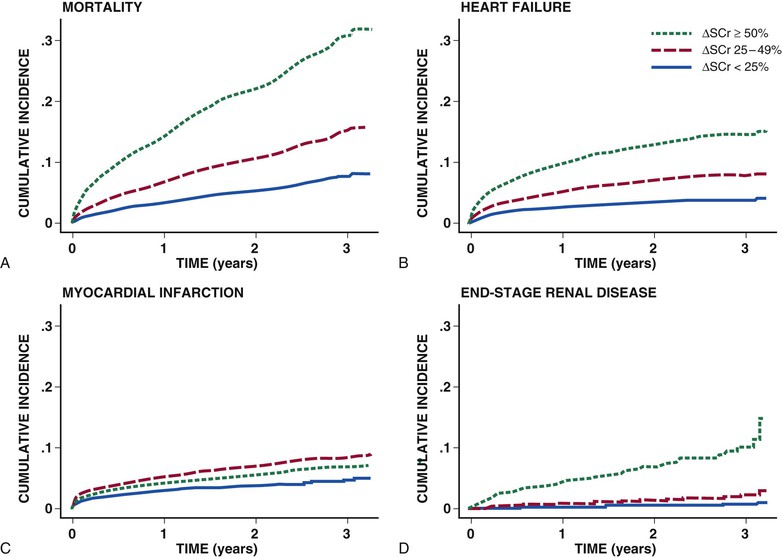
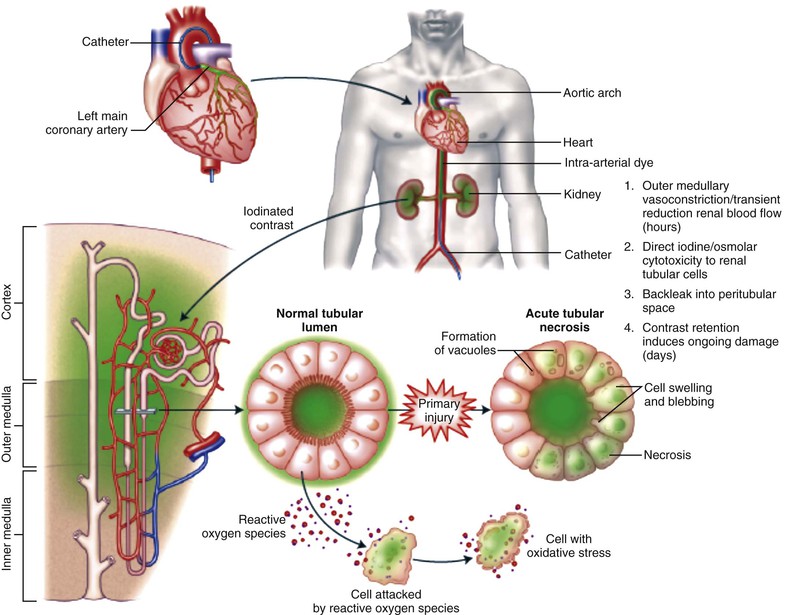
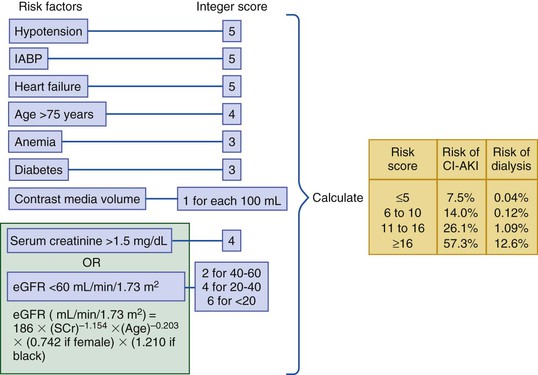
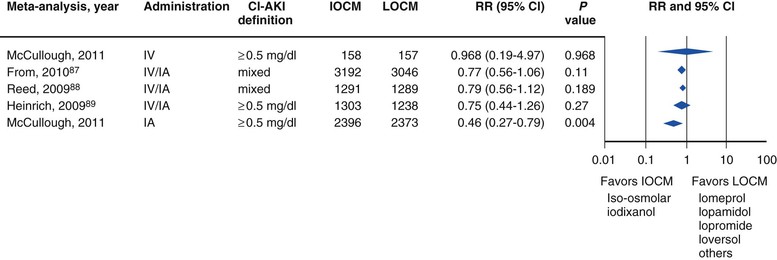
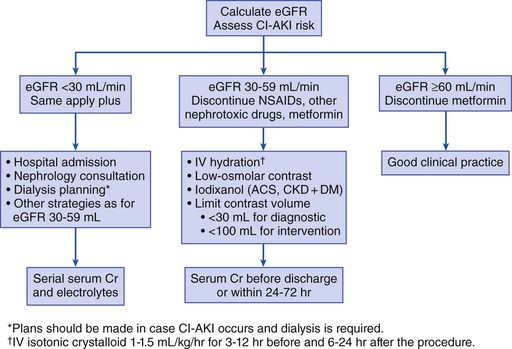
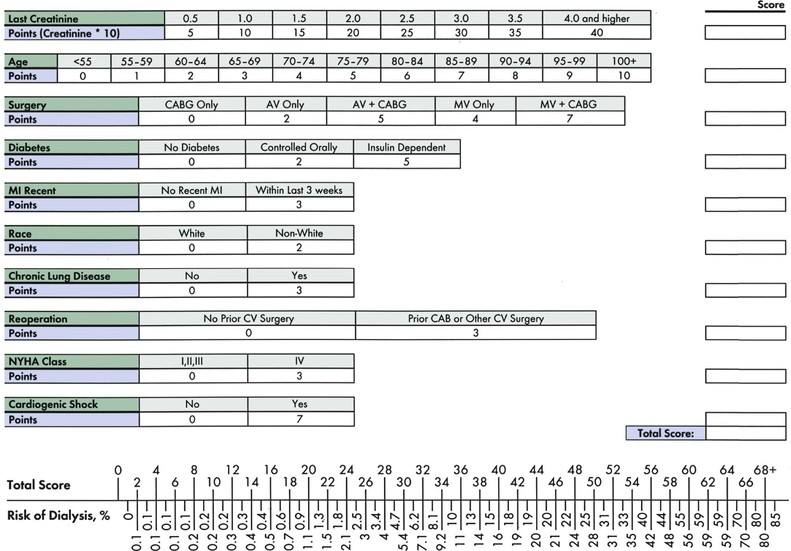
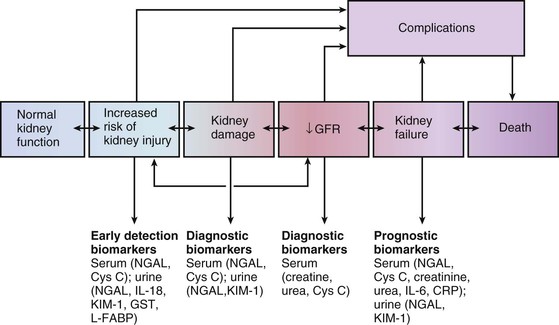
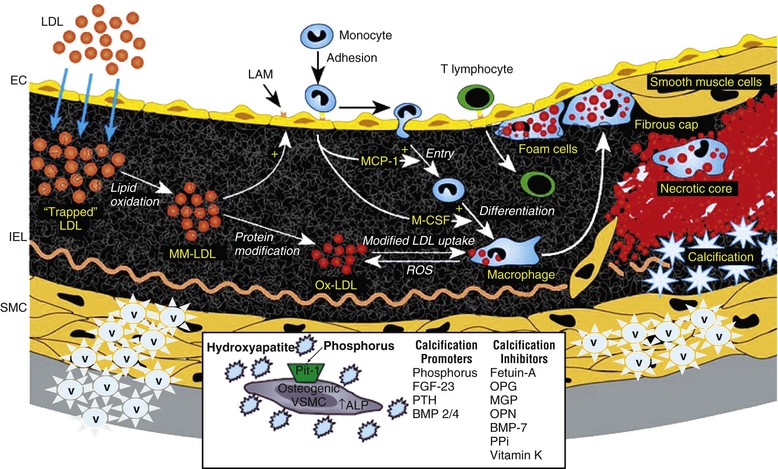
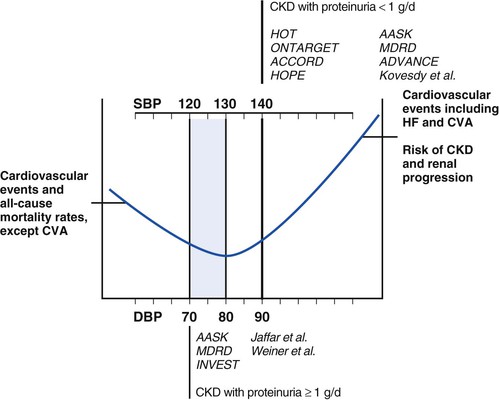
Peter A. McCullough The heart and kidney functions interrelate inextricably. The kidneys receive 20% to 25% of the cardiac output, perfusing approximately 1 million nephrons (Fig. 88-1). This flow per unit weight exceeds by several-fold that of most other organs. The kidney has a central role in electrolyte balance, protein production and catabolism, and blood pressure regulation. Communication between the heart and the kidneys occurs at multiple levels, including the sympathetic nervous system (SNS), the renin-angiotensin-aldosterone system (RAAS), antidiuretic hormone, endothelin, and the natriuretic peptides. Elucidation of these pathways has led to development of some of the key diagnostic and therapeutic targets in contemporary cardiovascular medicine. The obesity pandemic contributes to type 2 diabetes mellitus and hypertension, conditions that predispose to chronic kidney disease (CKD) and cardiovascular disease (CVD).1 Of persons with diabetes for 25 years or longer, approximately 50% will develop diabetic nephropathy.2 Approximately half of all cases of end-stage renal disease (ESRD) result from diabetic nephropathy. The aging of the population can be expected to accentuate this problem. Considerable evidence suggests that CKD accelerates atherosclerosis, myocardial disease, and valvular disease and promotes an array of cardiac arrhythmias.3 There is increasing recognition of the association among blood hemoglobin (Hb) level, CKD, and CVD. The World Health Organization (WHO) definition of anemia is a Hb level below 13 g/dL in men and below 12 g/dL in women. Approximately 9% of the general adult population meets the definition of anemia at these levels. In general, anemia due to CKD is present in 20% of patients with stable coronary disease and 30% to 60% of patients with heart failure. Hence anemia is a common and easily identifiable potential cause of constitutional symptoms as well as a potential diagnostic and therapeutic target, particularly in children.12,13 Anemia contributes to multiple adverse outcomes, in part owing to decreased tissue oxygen delivery and utilization.13 Of the many factors that may cause anemia in patients with CKD, of particular note is a relative deficiency of erythropoietin-alpha (EPO), an erythrocyte-stimulating protein (ESP), which normally is produced by renal parenchymal cells in response to decrease in blood partial pressure of oxygen. Normal plasma EPO levels range between 10 and 30 IU/mL; during anemic periods, however, these levels may be elevated up to 100 IU/mL. Patients with CKD and heart failure appear to resist the effects of EPO. In addition, increased circulating levels of hepcidin, an inhibitor of the ferroportin receptor, impair iron absorption and utilization throughout the body including the bone marrow. These factors can work to directly reduce red cell production at the level of the bone marrow and further worsen the anemia. In a review, 28 of 29 large prospective studies of heart failure found anemia to predict mortality independently.14 As Hb drops over time, a graded in increase in heart failure–related hospitalizations and death is seen. Conversely, those patients who have shown a spontaneous rise in Hb have fewer future events. This improvement is associated with a significant reduction in left ventricular mass index, suggesting a favorable change in left ventricular remodeling.15 Treatment of anemia with exogenous ESPs (EPO and darbepoetin alfa increasing the Hb level from below 10 g/dL to 12 g/dL can improve left ventricular remodeling, ejection fraction, and functional classification and raise levels of peak oxygen consumption with exercise testing. Yet treatment with EPO and supplemental iron (required in approximately 70% of cases), can cause three problems: (1) increased platelet activity, thrombin generation, and resultant increased risk of thrombosis; (2) elevated endothelin and asymmetric dimethylarginine which theoretically reduces nitric oxide availability, and results in hypertension; and (3) worsened measures of oxidative stress. Three randomized trials in CKD found that treatment with ESP’s resulted in higher CVD event rate. The Cardiovascular Risk Reduction by Early Anemic Treatment with Epoetin beta in Chronic Kidney Disease Patients (CREATE) Trial randomly assigned 600 patients to treatment with EPO to a target Hb of 13.0 to 15.0 versus 10.5 to 11.5 g/dL over 2.5 years and found higher rates of CVD and progression to ESRD in the 13.0 to 15.0 g/dL group.16 The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial randomly assigned 1432 patient with CKD and treated with EPO to groups with a target of 13.5 g/dL or 11.3 g/dL.17 The composite endpoint of stroke, MI, heart failure-related hospitalization, and death occurred in 125 subjects in the 13.5 g/dL arm and in 97 in the 11.3 g/dL arm (P = 0.03). The Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) was a multicenter, double-blind, placebo-controlled study in 4038 patients with CKD (eGFR, 20 to 60 mL/min/1.73 m2), type 2 diabetes mellitus, and anemia (Hb <11 g/dL) that randomly assigned subjects to darbepoetin alfa to raise Hb to 13 g/dL versus placebo, with rescue therapy for Hb levels below 9 g/dL. Use of darbepoetin alfa was associated with higher rates of fatal or nonfatal stroke (hazard ratio [HR], 1.92; P <0.001), with no differences in rates of ESRD or death.18 Thus treatment of anemia with ESPs may actually worsen CVD outcomes in CKD. Therefore ESPs should be reserved for the treatment of severe anemia in CKD with the goal of improving symptoms and avoiding transfusion. Iodinated CI-AKI is most commonly defined by the Acute Kidney Injury Network criteria of a 0.3 (mg/dL) or greater rise in serum creatinine from baseline within 48 hours of intravascular administration.19 CI-AKI occurs in approximately 13% in nondiabetics and 20% of those with diabetes mellitus undergoing PCI. The risk of CI-AKI relates in a curvilinear fashion to declining eGFR.20 Fortunately, among patients undergoing PCI, CI-AKI leading to dialysis is rare (0.5% to 2.0%). Nevertheless, severe CI-AKI is associated with catastrophic outcomes including a 36% in-hospital mortality rate and a 2-year survival rate of only 19%.21 Transient rises in serum creatinine are associated with longer length of hospital stay, MI, stroke, heart failure, and rehospitalization, among other complications22 (Fig. 88-4). Three core elements participate in the pathophysiology of CI-AKI: (1) direct toxicity of iodinated contrast material to nephrons, (2) microshowers of atheroemboli to the kidneys (consequent to catheter and wire exchanges above the renal arteries), and (3) contrast material– and atheroemboli-induced intrarenal vasoconstriction of the vasa recta. Direct toxicity to nephrons with iodinated contrast media appears to be related to the ionicity and osmolality of the contrast media.23,24 Microshowers of cholesterol emboli may contribute to AKI after PCI.25 Most of these showers are clinically silent. In approximately 1% of high-risk cases, however, an acute cholesterol embolism syndrome can develop, manifested by acute renal failure, mesenteric ischemia, decreased microcirculation to the extremities, and, in some cases, embolic stroke (see Chapter 58). Because acute renal failure occurs after coronary artery bypass surgery with nearly the same risk predictors as CI-AKI, atheroembolism may contribute to both causes of renal failure.26–29 Hypoxia triggers activation of the renal SNS, further reducing renal blood flow (Fig. 88-5). Preexisting renal dysfunction leads as a predictor of CI-AKI. When the eGFR falls below 60 mL/min/1.73 m2, the remaining nephrons must assume the residual filtration load, with increased oxygen demands and greater susceptibility to cytotoxic, ischemic, and oxidative injury.30 Patients with baseline eGFR < 60 mL/min/1.73 m2, in particular, those with CKD and diabetes mellitus, should receive preventive measures for CI-AKI. CKD, diabetes mellitus, and other risk factors including hemodynamic instability, use of intra-aortic balloon counterpulsation, heart failure, older age, and anemia in the same patient can produce a predicted probability of CI-AKI of greater than 50%31 (Fig. 88-6). Thus obtaining informed consent for contrast procedures from patients at high risk for CI-AKI should include disclosure of this possibility. Four clinical issues merit consideration in CI-AKI prevention: (1) hydration and volume expansion, (2) choice and quantity of contrast material, (3) pre-, intra-, and postprocedural end-organ protection with pharmacotherapy, and (4) postprocedural monitoring and expectant care. Hydration with intravenous normal saline or isotonic sodium bicarbonate is reasonable, starting 3 to 12 hours before the procedure at a rate of 1 to 2 mL/kg/hr.32–34 Patients at risk and able to tolerate the fluid load should receive at least 300 to 500 mL of intravenous hydration fluid before contrast administration. The largest trial (N = 381) of intravenous saline versus isotonic bicarbonate in elective angiography and PCI showed no differences in rates of CI-AKI (5.3% for saline, 9.0% for bicarbonate) or ESRD. Right-heart catheterization may aid management during and after the procedure for patients with heart failure. The postprocedure hydration target is a urine output of 150 mL/hr. Patients with urinary excretion rates of more than 150 mL/hr should have intravenous replacement of extra losses.33 This strategy generally calls for normal saline or sodium bicarbonate at 150 mL/hr for at least 6 hours after the procedure. Randomized trials of iodinated contrast agents have demonstrated the lowest rates of CI-AKI with nonionic, iso-osmolar iodixanol. A meta-analysis restricted to 25 head-to-head, prospective, double-blind, randomized, controlled trials that compared iodixanol with low-osmolar contrast media (LOCM) in adult patients undergoing angiographic examinations with serum creatinine values at baseline and after contrast administration.35 The relative risk of CI-AKI (creatinine rise ≥0.5 mg/dL) occurring for iodixanol was 0.46 (P = 0.004), compared with LOCM, as summarized in Figure 88-7. These data support the hypothesis that iodixanol (290 mOsm/kg) is less nephrotoxic than LOCM agents with osmolalities ranging from 600 to 800 mOsm/kg when the intra-arterial route of administration is used. With intravenous administration, however, rates of CI-AKI do not differ between iso-osmolar contrast media (IOCM) and LOCM.35 Contrast volume should be minimized in any setting, and there is disagreement about a “safe” contrast limit. The lower the eGFR, the smaller the amount of contrast material needed to cause CI-AKI. Maximum target volumes of contrast medium should be less than 30 mL for a diagnostic and less than 100 mL for an interventional procedure. With staged procedures, more than 10 days should elapse between the first and second contrast exposures if CI-AKI has occurred with the first procedure. A majority of trials of preventive strategies for CI-AKI have been small and underpowered and did not find the preventive strategy under investigation to be better than placebo. These trials have yielded a few lessons: (1) loop diuretics or mannitol can worsen CI-AKI if volume replacement is inadequate for the diuresis that follows; (2) low-dose or “renal dose” dopamine or fenoldopam does not provide protection despite the popularity of this approach in clinical practice, given the counterbalancing forces of intrarenal vasodilation through the dopamine-1 receptor and the vasoconstricting forces of the dopamine-2, alpha, and beta receptors; and (3) renal toxic agents including nonsteroidal anti-inflammatory agents, aminoglycosides, and cyclosporine should not be administered in the periprocedural period. After many small, suggestive studies, a large (N = 2308) randomized trial of N-acetylcysteine 1200 mg by mouth twice daily the day before and after the procedure showed no differences in the rates of CI-AKI (12.7% for both groups), ESRD, or other outcomes.36 No agent has received U.S. Food and Drug Administration approval for the prevention of CI-AKI. Figure 88-8 shows an algorithm for risk stratification and prevention of CI-AKI. It suggests for eGFR less than 60 mL/min/1.73 m2 use of optimal hydration, iodixanol or LOCM as the contrast agent, and a concerted effort to minimize the contrast volume.37 Postprocedural monitoring is critical in the current era of short stays and outpatient procedures. Ideally, in hospitalized high-risk patients, hydration should be started 12 hours before the procedure and continued for at least 6 hours afterward, with measurement of serum creatinine 24 hours after the procedure. Outpatients, particularly those with eGFR less than 60 mL/min/1.73 m2, either are admitted for an overnight stay or can be discharged to home with 48-hour follow-up and serum creatinine measurement. Patients who will develop severe CI-AKI usually show a rise of creatinine greater than 0.5 mg/dL in the first 24 postoperative hours. Thus discharge to home for those with less serum creatinine elevation and an otherwise uncomplicated course may be considered. Those with eGFR less than 30 mL/min/1.73 m2 require a discussion of the possibility of dialysis and a nephrology consultation regarding possible pre- and postprocedure hemofiltration and dialysis management.38 The kidney regulates blood pressure and controls intraglomerular pressure through autoregulation. Sodium retention stimulates increases in systemic and renal arteriolar pressure in an attempt to force greater degrees of filtration in the glomerulus. Glomerular injury activates several pathways that can increase systemic blood pressure. This effect sets up a vicious circle of more glomerular and tubulointerstitial injury and worsened hypertension (see also Chapters 43 and 44). A cornerstone of management of combined CKD and CVD is strict blood pressure control with a target of less than 130/80 mm Hg (Fig. 88-13). Most patients with CKD and proteinuria require three or more antihypertensive agents to achieve this goal blood pressure.48 Lifestyle management priorities for CKD and hypertension include dietary changes with sodium restriction, weight reduction of 15% or more to a target body mass index less than 25 kg/m2, and exercise for 60 minutes/day most days of the week. Pharmacologic therapy aims for strict blood pressure control with an agent that antagonizes the RAAS, often in combined action with a thiazide-type diuretic.48 Dihydropyridine calcium channel blocker monotherapy should be avoided because of relative afferent arteriolar dilation increased intraglomerular pressure and worsen glomerular injury. Combinations of multiple RAAS-blocking drugs—angiotensin-converting enzyme (ACE) inhibitor, angiotensin receptor blocker (ARB), direct renin inhibitor—have not shown superiority to single agents in this class and cause more complications. Clinical clues suggest underlying bilateral renal artery stenosis (e.g., poorly controlled blood pressure on more than three agents, abdominal bruits, smoking history, peripheral arterial disease, or a marked change in serum creatinine with administration of ACE inhibitor or ARB).49 Although renal artery stenosis accounts for less than 3% of ESRD cases, it represents a potentially treatable condition.50 Diagnostic and advanced therapeutic approaches for renal artery stenosis are discussed elsewhere in this text. Percutaneous renal artery denervation is undergoing evaluation as an approach to, lessen need for antihypertensive agents, and may improve cardiovascular outcomes (see Chapters 43, 44, and 60).51 Patients with CKD have higher rates of silent ischemia, risk of serious arrhythmias, heart failure, and other cardiac complications. In stable outpatients with CKD, 38% and 68% will have a high-sensitivity cardiac troponin I (cTnI) and cardiac troponin T (cTnT) above the 99th percentile of normal, respectively. The degree of elevation of cTn reflects left ventricular mass and the severity of renal disease.52 The cTnI predicts mortality in patients with ESRD.53 In general, cTnI serves best for diagnostic evaluation of patients with CKD or ESRD experiencing acute chest discomfort, whereas chronic elevations of cTnT are more common and more prognostic in stable patients. A rise and fall of cTnI or cTnT above the 99th percentile aids in the diagnosis of AMI in patients with CKD or ESRD. The skeletal myopathy of CKD can elevate creatine kinase, myoglobin, and some older-generation cTnI/cTnT assays, making these tests less desirable. Beyond biomarkers, the diagnosis of AMI should include confirmation of characteristic chest pain, electrocardiographic changes (ST-segment elevation or depression, new Q waves), or perfusion abnormalities or the identification of a culprit lesion on angiography.
Interface Between Renal Disease and Cardiovascular Illness
The Cardiorenal Intersection
Implications of Anemia Due to Chronic Kidney Disease
Contrast-Induced Acute Kidney Injury
Definition and Pathophysiology
Prevention of Contrast-Induced Acute Kidney Injury
Renal Disease and Hypertension
Diagnosis of Acute Coronary Syndromes in Patients with Chronic Kidney Disease
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Interface Between Renal Disease and Cardiovascular Illness
88