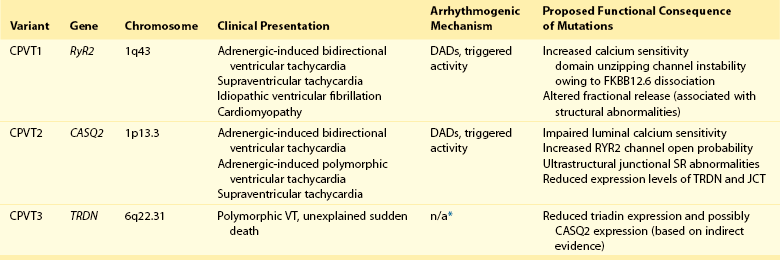
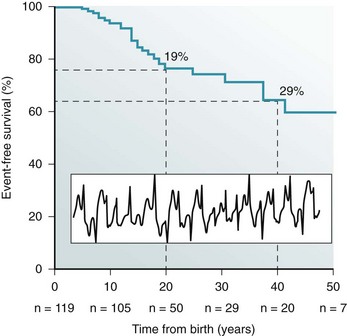
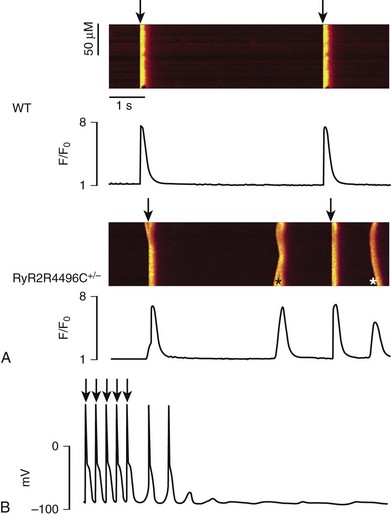
53 Growing clinical and experimental evidence highlights the relevance of cardiac calcium handling in the pathogenesis of inherited arrhythmias.1 The control of Ca2+ fluxes in myocardial cells requires the timely coordination of several events that ultimately lead to contraction. Any perturbation of this process has the potential to determine an arrhythmogenic substrate. The core events of Ca2+ ion movements in the myocardial cells are the opening of the L-type Ca2+ channels followed by the release of Ca2+ from the sarcoplasmic reticulum (SR) through the opening of the ryanodine receptors (RyR2).2 This process is called calcium-induced calcium release (CICR).3 The L-type calcium channel (CaV1.2) activation is therefore the first step of CICR. CaV1.2 belongs to the family of the voltage-gated calcium channels that are macromolecular complexes consisting of an ion conducting protein (the α1-subunit) and additional accessory peptides with regulatory function called α2δ, the β1-4, and γ subunits.4 Among the biophysical properties of voltage-dependent calcium current (ICa), the inactivation process is relevant to inherited arrhythmias (Timothy syndrome, see Chapter 95). Two components have been identified: voltage- (VDI) and Ca2+ (CDI)-dependent inactivation. The Ca2+-mediated component of inactivation is modulated by intracellular (cytosolic) concentration of calcium (Ca2+). CaV1.2 channels tend to cluster in the T-tubules in close proximity with the ryanodine receptors sitting across the membrane of the SR.5 CaV1.2 activation constitutes the signal for the activation of the RyR2. This latter event can be detected as a calcium transient, which is the sum of coordinated local releases that occur at specialized structures: the calcium release units (CRUs; Figure 53-1). One CRU is formed by clusters of RyR2 receptors that are in close proximity to L-type Ca2+ channels in the T-tubules.6 CRUs also include cardiac calsequestrin (CASQ2), triadin (TRDN), and junctin (JTC) that contribute to the control of the calcium release process. These peptides form a macromolecular complex that acts in coordination to control Ca2+ release. The number of CRUs recruited for release at each cardiac cycle is an important modulator of the systolic Ca2+ transient amplitude,7,8 and the loss of integrity of CRU is part of the pathophysiology of CPVT.9 Figure 53-1 Schematic representation of CRU and CPVT gene localization. The figure depicts a schematic representation of a T-tubule with the LTCC sitting across the plasmalemma juxtaposed to the RyR2/CASQ2/TRND/JTC macromolecular complex from the SR side. The SR component of CRU is involved in CPVT pathogenesis. CASQ2, cardiac calsequestrin; CPVT, catecholaminergic polymorphic ventricular tachycardia; CRU, calcium release unit; JTC, junctin; LTCC, L-type calcium channel; RyR2, cardiac ryanodine receptor; SR, sarcoplasmic reticulum; TRDN, triadin. During the relaxation phase, SR Ca2+ release terminates, and Ca2+ is taken up in the SR by the SR Ca2+-ATPase (SERCA) or extruded from the cell by the Na+/Ca2+ exchanger (NCX).10 NCX extrudes one Ca2+ ion (two positive charges) for every three Na+ ions (three positive charges) that are transferred into the cell. Thus, NCX generates a net inward depolarizing current, the transient inward current—Iti. In physiological conditions, SERCA is responsible for approximately 63% of calcium removal, and NCX mediates the remaining 37%.11 However, NCX becomes important to remove Ca2+ in any condition of calcium overload (e.g., in patients with genetic mutations of the RyR2 gene [CPVT] and during heart failure).11 Excessive activation of NCX can be arrhythmogenic. Activation of the adrenergic nervous system, mainly through β-adrenergic receptors, has profound effects on calcium handling, and it is often the initiator of calcium-mediated arrhythmogenesis.12 Adrenergic activation has two major effects on calcium handling: the enhancement of the amplitude of the L-type calcium current (ICa) and the increase of SR Ca2+ levels induced through the activation of SERCA13. The latter is responsible for an increase of Ca2+-transient amplitude. The effects of adrenergic activation can take place because of the phosphorylation target proteins14 induced by the activation of two enzymes with kinase activity: protein kinase A14 and Ca2+ calmodulin kinase II (CAMKII).15 Among the several target proteins, the adrenergic-dependent changes of Ca2+ handling are mainly due to the phosphorylation of L-type Ca2+ channel (increased current amplitude) phospholamban (removal of SERCA inhibition and increase of SR Ca reuptake) and RyR2 (increased open probability and increased transients).16 Thus, protein phosphorylation is an important mechanism that enables adrenergic activation to enhance the SR calcium release. In physiological conditions, this response is useful to react to environmental stressors by improving myocardial contractility. CPVT is the most frequent phenotype associated with mutations involved in intracellular calcium handling. Three genes have been implicated in its pathogenesis (Table 53-1): ryanodine receptor (RyR2 [CPVT1]),17 cardiac calsequestrin (CASQ2 [CPVT2]),18 and cardiac triadin (TRDN [CPVT3]).19 CPVT1 is an autosomal dominant trait, whereas CPVT2 and CPVT3 are rare autosomal recessive disorders. In 2001, Priori et al17 identified RyR2 as the gene responsible for the most frequent variant of CPVT. The prevalence of RyR2 mutations in patients with a clearly diagnostic phenotype is high (≈60% to 70%).20,21 Mutations concentrate in three specific areas of the RyR2 protein (amino acids 77-466, 2246-2534, and 3778-4967); however, 14% of CPVT patients harbor RyR2 mutations located outside these areas.21 Recent data suggest that there is no significant difference of outcome of CPVT according to mutation site, including the outcome of the subgroup with mutations outside the canonical clusters.22 N, RyR2 mutations have been reported also in patients referred for idiopathic ventricular fibrillation and in relatives of subjects who died suddenly.23,24 Autosomal recessive CPVT is rarely identified in the clinical setting. Combined estimated prevalence of the two recessive variants is approximately 5%. CPVT is a severe inherited arrhythmogenic disease manifesting with adrenergically mediated arrhythmias, often leading to syncope or cardiac arrest.17,23,25–27 Although the resting electrocardiogram is unremarkable, the reproducible inducibility of ventricular tachycardia (VT) during exercise stress test is the hallmark of the disease. Most patients with CPVT show a bidirectional VT pattern characterized by beat-to-beat 180-degree rotation of the QRS axis (Figure 53-2)17,23,26; however, polymorphic tachycardia or ventricular fibrillation may also be part of the picture.23,27 This unstable, catecholamine-sensitive, substrate can lead to sudden death as the first manifestation of the disease in up to 30% of cases.23 Symptoms suggesting the presence of arrhythmias tend to manifest early in life (median age, 12 years), although later onset is possible. The development of palpitations and syncopal events during adrenergic stress is a critical element to suspect the diagnosis of CPVT, which is confirmed by the induction of bidirectional VT on an exercise stress test. In untreated patients, the occurrence of severe arrhythmias is approximately 60% to 70%, and approximately 30% experience a cardiac arrest or sudden death upon first manifestation (see Figure 53-2).23,28 CPVT patients typically present with structurally normal heart. However, some RyR2 mutations have been associated anecdotally with structural cardiomyopathies. One of the first reports on RyR2 gene mutations suggested an association with arrhythmogenic right ventricular cardiomyopathy (ARVC).29 More recently, other authors have reported preliminary data linking RyR2 with hypertrophic cardiomyopathy (HCM).30 Although careful scrutiny of these clinical reports does not establish a causal link, recent experimental findings suggest a mechanistic explanation as to why some RyR2 variants could in principle make the heart more prone to develop structural abnormalities (discussed later). Figure 53-2 Kaplan-Maier curve showing the cumulative occurrence of cardiac arrest, sudden death, or ICD shocks in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) in the absence of therapy. An example of bidirectional ventricular tachycardia, the typical CPVT arrhythmia, is depicted in the inset. β-Adrenergic receptor block represents the first-choice therapeutic approach. The use of β-blockers is based on the evidence of the direct link between adrenergic activation and cardiac events in CPVT. Clinical data show that β-blockers have an effect on the natural history of CPVT by achieving a significant reduction of cardiac events. In this context, exercise stress testing is important for dosage titration because the threshold for arrhythmias in CPVT is reproducible. β-Blockers can prevent the onset of arrhythmia in 70% to 80% of cases.23,26 In addition, nonselective β-blockers (e.g., nadolol, propranolol) confer the highest degree of protection against the onset of VT and SCD. However, the nontrivial incidence of recurrent cardiac on optimal β-blocker dosage (approximately 30%)22 calls for the identification of additional therapeutic strategies. Implantable cardioverter defibrillator (ICD) implant, left cardiac sympathetic denervation, and other pharmacologic approaches have been proposed, and flecainide appears to be the most promising strategy. Although the mechanisms for its effectiveness are debatable (discussed later), clinical data and personal experience suggest that flecainide affords additional protection when added to β-blockers in CPVT.31,32 From a mechanistic standpoint, similarities and significant differences exist among CPVT1-3 genetic variants. In all three forms of CPVT, arrhythmogenesis is a consequence of the occurrence of spontaneous calcium release (SCR; Figure 53-3). This term is used to refer to SR Ca2+ release events that are not driven by a stimulated action potential. Whenever an SCR occurs and the levels of Ca2+ in the sarcolemma increase, the NCX activates to extrude the excess of ions. As mentioned earlier, NCX activation generates the Iti current, which is the cause of the development of delayed afterdepolarizations (DADs)33 and transient membrane depolarizations occurring during phase 4 of the action potential (see Figure 53-3). When DAD amplitude reaches the voltage threshold for sodium channel activation, triggered beats occur. Figure 53-3 A, Representative line-scan images and Ca2+ transients in a wild type myocyte and a RyR2R4496C+/− myocyte at 0.2 Hz pacing in the presence of isoproterenol (100 nM). Asterisk indicates spontaneous Ca2+ release in the RyR2R4496C+/− myocytes. Arrows indicated the field stimulations. B, Action potential recording in a myocyte isolated from a RyR2R4496C+/− during exposure to isoproterenol 30 nmol. Arrows indicate stimulated beats, which are followed by two triggered beats and delayed afterdepolarizations. The effects of CPVT mutations have been analyzed in a variety of experimental settings, including lipid bilayer, heterologous expression systems, murine transgenic models, and in myocytes derived from patient-specific induced pluripotent stem cells.34 The following sections outline the key concepts about arrhythmogenesis in each form of CPVT. The RyR2 channel is a homotetramer; each subunit is formed by 4967 amino acids with a long (≈4300 amino acids) N-terminal cytoplasmic domain.35 The last 500 amino acids at the C-terminal of RyR2 form the transmembrane segments encircling the channel pore. The regulation of RyR2 opening and closing (gating) is mainly controlled by Ca2+ levels at cytoplasmic and luminal SR sides, and it is facilitated when the Ca2+ concentration at either side increases.36,37 Therefore, RyR2 calcium sensitivity is an important physiological function that controls CICR. The majority of RyR2 mutations identified in CPVT patients cause increased calcium sensitivity (defined as “gain-of-function”). Marks et al. showed that mutant RyR2 exhibits an increased sensitivity to cytosolic Ca2+ after protein kinase A phosphorylation. They also suggested that such change in sensitivity is due to an abnormal dissociation of FKBP12.6, a putative RyR2 stabilizing protein.38 During adrenergic stimulation, phosphorylation of RyR2 would promote SCRs by further dissociation of FKBP12.6 with a consequent excessive increase of open probability.
Inheritable Phenotypes Associated With Altered Intracellular Calcium Regulation
Overview of the Function of the Calcium-Handling System

Sarcoplasmic Reticulum Ca2+ Release Threshold and the Adrenergic Signaling
Phenotypes Associated With Mutation in Calcium Handling Proteins
Clinical Manifestations and Management of CPVT
Pathophysiology
Abnormalities of Calcium Handling in CPVT
Ryanodine Receptor, Mutations, and CPVT
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Inheritable Phenotypes Associated With Altered Intracellular Calcium Regulation