Idiopathic Pulmonary Fibrosis
OVERVIEW
In simplest terms, pulmonary fibrosis equates with the growth of “scar” in the lung. Scarred lung can assume any one of a variety of patterns; these patterns define unique pathologic phenotypes. When scar assumes a patchy distribution and appears to emanate from the pleural surface, it is recognized as the “usual” type—otherwise known as usual interstitial pneumonia (UIP). The disease in which UIP manifests without cause or trigger is called idiopathic pulmonary fibrosis (IPF). IPF is, therefore, a type of pulmonary fibrosis (also known as interstitial lung disease [ILD]). IPF is recognized by a unique compilation of clinical, radiographic, and pathologic features. IPF causes breathlessness, disability due to respiratory insufficiency and, in most instances, eventually leads to death.
While there are many causes of ILD, IPF is the most common form of ILD and certainly the most serious. IPF is characterized by an inexorable progression of interstitial pulmonary fibrosis that results in restrictive lung disease and worsening gas exchange. Death from respiratory failure is reported to ensue within 5 years of diagnosis in the majority of patients.
By definition, IPF is UIP in the absence of cause or explanation. It is equally important to remember what IPF is not. IPF is not pulmonary fibrosis from chronic aspiration, drug toxicities, environmental exposures (such as chronic hypersensitivity pneumonitis), and collagen vascular diseases, even if the pattern of fibrosis resembles UIP. Therefore, the diagnosis of IPF can only be made by excluding other possibilities. One of the major challenges in diagnosing IPF is the reasonable exclusion of other potential causes of pulmonary fibrosis. This requires a careful clinical and diagnostic evaluation; and often verges on areas of clinical uncertainty.
IPF typically comes to medical attention later in life, beginning in the sixth decade. IPF is rarely the cause of ILD in patients under the age of 40. The predominant presenting symptoms of IPF are exertional breathlessness and a dry, harassing cough. These are nonspecific complaints shared by a variety of pulmonary and cardiac diseases. In particular, exertional breathlessness is often attributed to advancing age by patients in their sixties and seventies, leading to delays in seeking medical evaluation. In addition, many patients are poorly conditioned and overweight and attribute their symptoms of breathlessness to these circumstances. In addition to nonspecific clinical symptoms, initial nonspecific radiographic findings also fail to trigger prompt medical evaluation. Fine peripheral linear radiographic opacities on plain chest radiographs, predominantly in the lower lung zones, may be interpreted as chronic and nonspecific pulmonary fibrosis, which often does not elicit an alarming response. Delay in diagnosis is therefore the norm. However, in recent years important scientific advances have improved the understanding of IPF pathogenesis; new therapeutic trials are under way and this has increased the enthusiasm for an early diagnosis of IPF.
HISTORICAL PERSPECTIVE
A brief review of the evolution in our understanding of IPF illustrates the contributions made by earlier investigators and account for much of the confusion that many clinicians have regarding IPF. One of the challenges in defining IPF has been the variety of antiquated terms formerly used to describe pulmonary fibrosis. While there are many causes of ILD in general, and pulmonary fibrosis in particular, it is important to note that IPF is a unique disease, although it had not been formally codified until recently when a group of expert pulmonologists, radiologists, and pathologists collaborated on a classification of ILD. Reviewing the history of IPF will both clarify the present terminology and distinguish contemporary nomenclature from the outmoded terms encountered in earlier literature.
Fibrosis of the lung was long recognized in association with infection or dust inhalation. In the 19th century, pulmonary fibrosis was known as “cirrhosis” of the lung. Yet little attention was paid to this form of respiratory illness.1 Interest in pulmonary fibrosis was ignited in 1944 when Drs. Louis Hamman and Arnold Rich published a seminal paper describing “acute diffuse interstitial fibrosis of the lungs.”2 Hamman and Rich reported a series of unusual cases that shared a unique clinical presentation featuring idiopathic subacute respiratory failure followed by death. Their report was complete with pathologic findings from autopsy. They described thickening of the alveolar interstitium and areas of dense fibrotic scar tissue within the lung. This was the first pathologic depiction of pulmonary fibrosis and, to this day, is considered an accurate portrayal. In retrospect, the cases of Hamman and Rich best fit a diagnosis of the fibrosing interstitial pneumonia now known as acute interstitial pneumonitis (AIP).3 Yet in the 1940s, the “Hamman–Rich syndrome” became synonymous with IPF. So it remained for the next three decades.
Over the years, clinical reports of pulmonary fibrosis suggested a number of alternate presentations that were referred to as “variants” of the Hamman–Rich syndrome.4 This included cases that exhibited a rather protracted duration of illness compared to the “classic” Hamman and Rich cases. It was also noted that pulmonary fibrosis occurred in patients who suffered from the “rheumatoid group of collagen diseases.” An assortment of abnormal patterns was noted under the microscope. Eventually, the breadth of the Hamman–Rich syndrome encompassed a heterogeneous mixture of clinical manifestations and a variety of histologic forms of pulmonary fibrosis with no distinction made between systemic and limited illness nor any concession to the prognostic implications of an acute versus chronic presentation.
In the 1960s authors began to regularly substitute the term “IPF” for “acute diffuse interstitial fibrosis.”5,6 A debate began concerning the chronicity of this disease, with some authors suggesting a slow course punctuated by “terminal complications,”5 while others reported an average illness of no more than 2 years.6
The term “fibrosing alveolitis” was introduced in England in 1964.7 Cryptogenic fibrosing alveolitis (CFA) became the preferred term for pulmonary fibrosis in the European literature and it is essentially synonymous with IPF. This term was originally meant to improve upon its predecessor by capturing pathologic features in a manner that was more precise and descriptive. CFA refers to the interalveolar location of the inflammation in pulmonary fibrosing as compared with the intra-alveolar inflammation of infectious pneumonia. This interalveolar septal inflammation was dubbed “alveolitis.” It was maintained that alveolitis was responsible for the subsequent development of fibrosis and it was first suggested that corticosteroids be used to treat alveolitis and therefore pulmonary fibrosis.
The most important advance came in 1964 with the publication of an improved and safe technique for performing open lung biopsy.8 With this procedure, it became possible to carry out a widespread analysis of lung tissue from patients with suspected pulmonary fibrosis. Before long there were new insights into the pathology associated with fibrotic lung disease.
In 1969 Liebow and Carrington9 heralded the modern era of ILD histopathology with the notion that idiopathic interstitial pneumonia (IIP) could be split into separate pathologic subtypes. They described distinct patterns of IIP that were identified by examination of lung biopsy specimens with light microscopy. Moreover, these subtypes were found to predict prognosis and response to treatment. Based on their research findings, Liebow and Carrington produced the first detailed histopathologic classification of IIP. They created five categories that were termed UIP, desquamative interstitial pneumonia (DIP), bronchiolitis obliterans interstitial pneumonia (BIP), lymphocytic interstitial pneumonitis (LIP), and giant cell interstitial pneumonia (GIP). More recent observations have led to a modification of this classification of IIP subtypes.10,11 New categories have been added, such as respiratory bronchiolitis–associated interstitial lung disease (RB-ILD) and nonspecific interstitial pneumonia (NSIP).10,11
Simultaneously, a revolution in thinking about the pathogenesis of IPF affected the way in which experts talked about the disease. Researchers at the National Heart, Lung, and Blood Institute (NHLBI) were major proponents of an “inflammatory theory” of pathogenesis as originally proposed by European investigators. This theory was based on studies at the NHLBI throughout the 1970s during which excessive amounts of inflammatory cells were identified in bronchoalveolar lavage (BAL) fluid obtained from IPF patients.12 The NHLBI agreed with European researchers who had coined the term “alveolitis” and the NHLBI also endorsed corticosteroid treatments. The inflammatory theory has since fallen from favor, mostly as a consequence of corticosteroid inefficacy, and the term “alveolitis” has also fallen out of vogue.
A new hypothesis has replaced the “inflammatory theory” of IPF. This new concept proposes that IPF is the result of alveolar epithelial injury, which is then followed by aberrant repair mechanisms. This theory emerged from landmark ultrastructural studies performed in the mid-1980s.13,14 Using electron microscopy, it was discovered that the alveolar epithelial cells were injured in IPF. In addition, foci of subepithelial fibrosis were first described. This concept of injury and repair was modified and expand on by subsequent investigators.15
In 1997 a modified version of Liebow’s pathologic classifications were proposed.16 The new classification scheme reinforced acceptance of certain categories within the context of an updated understanding of ILD pathogenesis. For instance DIP and UIP categories were retained in the new classification scheme. Some original categories were discarded and two modern categories were added. RB-ILD was recognized in the spectrum of smoking-related lung diseases and a provisional category, NSIP, was also added.16,17 This modern pathologic classification became the basis for a consensus statement that finally standardized the nomenclature of ILD and IPF for the very first time.
In 2002 a panel of experts convened sponsored jointly by the American Thoracic Society and the European Respiratory Society.18 This panel released an official statement for the purpose of providing a new and comprehensive classification of IIP that considered all clinical, radiographic, and pathologic features. The diseases recognized by the 2002 ATS/ERS classification of IIP are IPF, NSIP, cryptogenic organizing pneumonia (COP), acute interstitial pneumonia, RB-ILD, DIP, LIP. Further classification of IPF has been undertaken by joint consensus statements of the American Thoracic Society, and European Respiratory Society, in 2000 and 2010.19,20 These statements attempt to offer strict definitions for each subtype of IIP with practical guidelines for diagnostic purposes. The benefit of attempting to utilize precise definitions is to provide a uniformity of diagnostic decisions in both clinical practice and in research.
Despite the utility of rigorously defining IIP (and IPF), there are several potential pitfalls. For instance, the current classification system relies upon an assumption that each specific IIP is a discrete clinical entity. There has never been validation of this assumption by careful phenotyping and prospective trials. In addition, these definitions are heavily reliant on surgical pathology, suggesting that pathology be considered the “gold standard”; however, evidence shows even expert pathologists can have difficulty agreeing on pathologic classification.21 This continues to create confusion and difficulty in the diagnosis of IIPs and IPF. Hopefully continued refinement of consensus guidelines, greater expertise of clinicians and greater identification and use of genetic signatures/biomarkers will lead to more confident separation of the IIPs and a clear definition of IPF.
EPIDEMIOLOGY
Below are considered the incidence and prevalence of IPF, along with risk factors and associated familial and genetic factors.
 INCIDENCE, PREVALENCE, AND VITAL STATISTICS
INCIDENCE, PREVALENCE, AND VITAL STATISTICS
The epidemiology of IPF is difficult to determine and the available data are of limited value. It has been principally assessed by large population studies using death certificates and/or medical coding as the principle component for determination of disease. The main criticism of these studies is that surgical lung biopsy (SLB) was rarely performed or incorporated into the criteria used for analysis, although SLB remains the gold standard of diagnosis. In addition, many of these studies have not incorporated the up-to-date definition of the disease and use disparate methodologies to develop their assessments of the epidemiology of IPF.22,23 The most recent update of the diagnostic criteria for IPF may help to clarify these difficulties but will also make it difficult to compare data over time. Early studies from Great Britain and from the United States suggest that IPF is widely underreported.24,25 Though this is probably still the case heightened clinical awareness of the disease and the greater availability of high-resolution computed tomography (HRCT) appear to be changing the landscape; and this may explain recent studies that suggest an increasing incidence of the disease.22
The precise incidence and prevalence of IPF remains difficult to determine. However, there are several studies worth considering. Coultas et al.26 utilized a population-based registry in which cases were determined using a combination of medical records, pathology reports, and death certificates. With this data, the prevalence/annual incidence was estimated at 20.2/10.7 cases per 100,000 males and 13.2/7.4 per 100,000 females.
Recent studies by two different groups have attempted to update and further define these estimations.27,28 Both studies employed sensitivity analyses to examine the impact of diagnostic reliability; they examined the epidemiology of IPF using both narrow- and broad case definitions. Fernandez et al.27 reported a prevalence of 27.9 per 100,000 people, using a narrow case definition, and 63.0 per 100,000 people using a broad case definition; similarly, annual incidence was estimated at 8.8/100,000 and 17.43/100,000 using the narrow and broad definitions respectively. Raghu et al.28 relied on yet another North American database and reported a prevalence of 14.0 per 100,000 persons with their narrow definition and 42.7 per 100,000 persons with their broad case definition; annual incidence was reported as 6.8 and 16.3 per 100,000 persons for narrow and broad definitions respectively. It is important to point out that Fernandez et al. and Raghu et al. used different definitions of IPF in their separate studies, which likely accounts for some of the difference in the incidence and prevalence between the two studies. In European epidemiologic studies, there is even wider variation in case definition and reported incidence and prevalence; though some authors have claimed that IPF is more common in the United States than in Europe.23
Mortality data is equally difficult to determine, as data is scant and varies by country and race. Retrospective longitudinal studies have suggested that median survival is 2 to 3 years from time of diagnosis.29–33 However, new information is coming from the placebo arms of recent clinical trials. This data suggests that survival time may be greater than previously expected.34–36 As a result of this emerging data, the actual mortality of IPF therefore remains without clear definition.
 RISK FACTORS
RISK FACTORS
IPF remains a disease without known pathogenesis, which makes the definition of risk factors problematic. Despite this shortcoming, a few case-control observational studies have identified potential risk factors that include age, gender, smoking status, environmental exposures, gastroesophageal reflux, and viral infections. The identification of these risk factors remains just associations as research identifying causality is either ongoing or inconclusive. Despite the lack of clear causality these risk factors can help to identify patients who have higher risk of developing IPF.
Age/Gender
The incidence of IPF undoubtedly increases with age and appears to have a higher predilection for men. Patients with IPF are usually between 40 and 70 years old. Two-thirds of IPF cases present in patients over the age of 60 years, with a mean age of 66 years at the time of diagnosis.19 IPF occurs infrequently amongst those younger than 40 years and rarely affects children, if at all. Several studies stratified the incidence and prevalence of IPF by age.26–28,37,38 Amongst adults aged 35 to 44 years the prevalence was 2.7 cases per 100,000 persons. In contrast, the prevalence for individuals older than 75 years was greater than 175 cases per 100,000.26 Other studies in both USA and Europe have demonstrated similar findings.23 In addition, there appears to be a higher incidence and prevalence of IPF in males than in females with the notable exception of a study in Norway, which identified a higher incidence and prevalence in females.37
Smoking
Another risk factor that emerges from case-control studies is a history of cigarette smoking. The prevalence of tobacco use in patients with IPF is high, ranging from 41% to 83%.19,39 A meta-analysis of five case-control studies demonstrated that IPF patients were significantly more likely to report a history of smoking than controls with an odds ratio of 1.58 (95% CI 1.27–1.97).40 There may even be a dose–response relationship. Baumgartner et al.41 reported that IPF patients with a greater than 21-pack-year smoking history had an odds ratio of 2.26 (95% CI = 1.3–2.8) compared to individuals with less than a 20-pack-year history. This finding was corroborated in a subsequent study.42 Despite the association, a mechanistic link between smoking and IPF remains undefined.43
Environmental Exposures
A number of papers have implicated environmental exposures to such particulate materials as metal and wood dusts.20 In a related finding, an increased incidence of IPF was noted in industrial centers of the southeastern United States and central regions of the United Kingdom.44 There is also an association between farming and risk of IPF.40 A specific association exists between exposure to livestock and the risk of developing IPF though this seems to be strongest at exposures greater than 5 years.45 At this point these types of risk factors are only associations as the causative link to environmental exposures remains undefined.
Viral Infections
Several articles have implicated a variety of viruses such as the Epstein–Barr virus, influenza virus, cytomegalovirus, and hepatitis C.46–48 All are found with higher incidence amongst patients with IPF. The best studies are of herpes viruses (including EBV, CMV, human herpes virus 7 and 8). Tang et al.49 identified DNA from herpes virus in 33 patients with IPF. Herpes virus antigens have also been detected by immunohistochemistry in type II alveolar epithelial cells from IPF patients but were not seen in cells from patients with normal lungs.49–51 The significance of these findings remains unclear because there is still no evidence to support a pathogenic mechanism for IPF involving viruses. Interestingly, new animal models have shown a role for viral infection in the development of experimental fibrosis.52,53 Despite the suggestive evidence of virus in human IPF cells and potential mechanistic links from animal modeling, direct causal links to human disease remain undefined.
 FAMILIAL AND GENETIC FACTORS
FAMILIAL AND GENETIC FACTORS
Familial cases of IPF have been described in dozens of reports. The clinical features of familial IPF are indistinguishable from those of the nonfamilial form, except that the familial form may have an earlier age of onset.54 Familial IPF or familial interstitial pneumonia (FIP) is defined by at least two members of a primary biologic family (parent, child, siblings) presenting with a characteristic appearance of IPF that is confirmed by biopsy. Familial IPF seems to account for 0.5% to 2% of all cases of IPF.55
In 2000, a report was published describing 25 families and comprising 67 cases of familial IPF.55 In this report the mean age at time of diagnosis was 56 years. Only half of the patients were smokers. The male-to-female ratio was 2:1 in contrast to earlier reviews of FIP, which suggested an inverted male-to-female ratio. This was followed by a larger study of FIP published in 2005.56 This impressive report described a much larger cohort of 111 families with 309 affected family members. Most of these subjects were identified as having probable or definite IPF by the American Thoracic Society/European Respiratory Society diagnostic criteria. Interestingly, correlation with biopsy specimens suggested that FIP could present with a variety of pathologic patterns (other than UIP), even within the same family. This study revealed a mean age at diagnosis of 68.3 years, with a slight male predominance (55%) and an increased association with cigarette smoking (even after controlling for age and gender differences). Analysis of pedigrees confirmed vertical transmission and provided strong evidence for an autosomal dominant inheritance pattern of disease with variable penetrance.
These accounts of FIP provide compelling evidence for genetic factors that predispose to the development of IPF. One such factor is a mutation in the gene that encodes surfactant protein C (SPC) gene.57 Other studies have implicated mutations in the gene for surfactant protein A; and mutations in the genes that encode telomerase reverse transcriptase and the telomerase RNA template.58,59 By leveraging genomic linkage, investigators have recently described a common polymorphism in the promoter of MUC5B, which is associated with both FIP and sporadic IPF.60 This was followed by a large genome-wide association study of patients with IIPs, which identified additional genetic loci suggesting potential genetic markers of disease.61 This data is opening up insights into potential genetic pathways of this disease and confirms a generally understood concept that there is a combination of genetic factors and environmental exposures, which account for the significant disease heterogeneity observed in individual patients. While these studies provide useful insights they do not define a direct causal link at present. Several candidate genes have been selected, because of their bearing on proposed mechanisms of the disease, and these genes are currently under investigation.
CLINICAL PRESENTATION
In this section, we discuss important aspects of the clinical presentation of IPF.
 DIAGNOSIS
DIAGNOSIS
Differential
In the setting of exertional breathlessness, the hallmark of IPF is a predominance of radiographically visualized lower lung zone reticular opacities that spread out over time to involve an ever enlarging area of lung parenchyma (Fig. 56-1). The differential diagnosis of IPF includes the other IIP, connective tissue diseases (principally scleroderma and rheumatoid arthritis), chronic hypersensitivity pneumonitis, environmental exposures, occupational exposures, chronic aspiration, and heritable conditions such as the Hermansky–Pudlak syndrome. The aforementioned disorders all present with exertional dyspnea coupled with radiographic abnormalities indicative of an interstitial pulmonary disorder.
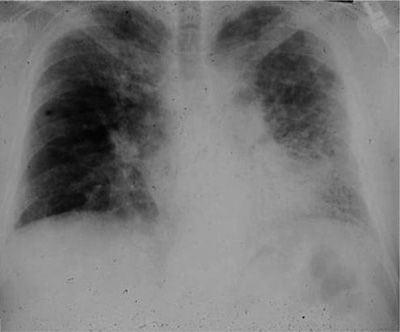
Figure 56-1 Posteroanterior chest radiograph of a 67-year-old man with progressive dyspnea revealing bilateral reticular infiltrates with lower lobe predominance.
HRCT has emerged as the single most important diagnostic modality in ILD. A number of diseases share a radiographic pattern that is similar to IPF, in other words, reticular abnormalities are demonstrated by HRCT with a tendency to involve the lower lobes. Examples include asbestosis, chronic aspiration, radiation pneumonitis, chronic hypersensitivity pneumonitis, end-stage sarcoidosis, and congenital disorders such as Gaucher disease, Niemann–Pick disease, and tuberous sclerosis–lymphangioleiomyomatosis. The presence of extensive ground-glass opacities on HRCT should prompt the consideration of an alternative diagnosis such as DIP, cellular NSIP, or acute hypersensitivity pneumonitis. Other IIP that are included in the differential diagnosis of IPF are fibrotic NSIP and COP.
History
Patients with IPF typically present with exertional dyspnea and a nonproductive cough. The dyspnea begins insidiously and is usually progressive. Dyspnea is the most prominent symptom in IPF. Associated systemic symptoms can occur but are not common. Systemic symptoms may include weight loss, low-grade fevers, fatigue, arthralgias, or myalgias.
Patients will often have symptoms for longer than 6 months before seeking medical attention. It is not unusual for symptoms to be present for up to 2 years before an initial consultation is arranged with a pulmonary specialist. Patients are frequently evaluated and treated for other ailments, such as asthma or heart failure, before IPF is identified as the final diagnosis. Because most patients present over the age of 60 where coronary artery disease is highly prevalent, many patients are referred for a cardiac evaluation before pursuing a pulmonary evaluation.
The patient’s age is an essential clue to the recognition of IPF. While IPF mostly occurs in older patients (>50 years), the other ILDs are more common among the young or middle-aged (examples include sarcoidosis, lymphangioleiomyomatosis, and pulmonary Langerhans cell histiocytosis).
A history of cigarette smoking is a vital piece of information. While IPF, DIP, and PLCH are diseases found in former and current smokers, other diseases such as hypersensitivity pneumonitis are rare among the smoking population.
It is critical to obtain a detailed occupational history with particular attention to exposures such as asbestos, silica, or any other respiratory toxins. This history is necessary to exclude the presence of pneumoconiosis. It is equally important to inquire about exposure to molds and/or pets in the home environment as this information may suggest a diagnosis of hypersensitivity pneumonitis.
A general health history, including an accounting of all medications, can be revealing. A review of systems may uncover photosensitivity, Raynaud phenomenon, dry eyes, or dry mouth that implies a connective tissue disorder. Certain drugs have been associated with pulmonary fibrosis, most notably nitrofurantoin, bleomycin, and amiodarone.
Physical Examination
In most patients the physical examination reveals fine, bibasilar inspiratory crackles, known as “Velcro rales.” As the disease progresses, rales can extend toward the upper lung zones. Clubbing is found in up to 50% of patients with IPF. Resting arterial oxygen saturation may be normal but desaturation is expected with exercise. Extrapulmonary involvement does not occur in IPF. Thus the physical examination is otherwise unremarkable in the early stages of the disease.
Later in the course of disease weight loss, cyanosis, and signs of pulmonary hypertension with cor pulmonale may become apparent. Findings at this stage include an accentuated pulmonic second heart sound, presence of a third heart sound, a right ventricular heave, and edema of the lower extremities.
Routine Laboratories
A routine laboratory evaluation is not helpful except for its role in ruling out other causes of diffuse parenchymal lung disease. Polycythemia is a rare finding despite the frequency of chronic hypoxemia. Elevation of systemic inflammatory markers (i.e., erythrocyte sedimentation rate or C-reactive protein level) or the presence of hypergammaglobulinemia is found in IPF yet such findings are nondiagnostic. The lactate dehydrogenase activity is often elevated but is also nonspecific. Up to 30% of patients with IPF may have positive tests for antinuclear antibodies or rheumatoid factor. These titers are not generally high. The presence of a high titer of autoantibodies suggests connective tissue disease while an elevated angiotensin-converting enzyme level or antineutrophil cytoplasmic antibodies indicates alternative diagnoses.
 PULMONARY FUNCTION AND PHYSIOLOGY
PULMONARY FUNCTION AND PHYSIOLOGY
Pulmonary function tests in IPF normally identify a restrictive ventilatory defect with reductions of total lung capacity (TLC), functional residual capacity (FRC), and the residual volume (RV). These changes are the result of diminished lung compliance. Pressure–volume studies will yield a curve that is shifted downward and to the right, indicative of lost lung compliance. As the disease progresses, compliance decreases further. Forced expiratory volume in 1 second (FEV1) and forced vital capacity (FVC) will also be decreased.
Unless a complicating airways disease is present (e.g., chronic obstructive pulmonary disease [COPD]), isovolume flow rates are preserved. While functional alterations associated with small airways disease have been reported in IPF, this description is exclusive to smokers and likely represents a concurrent smoking-related airways disorder.62
Impaired gas exchange is demonstrated by the measurement of a lowered diffusing capacity. The decline of diffusion capacity may even precede the development of abnormal lung volumes. Resting arterial blood gases are usually normal in IPF or else they will reveal mild hypoxemia with a respiratory alkalosis. Patients with IPF have tachypnea and often develop a pattern of rapid-shallow breathing. The work of breathing is increased in IPF. While no chemical changes can explain the observed hyperventilation, it is felt that rapid respiratory rates are secondary to altered mechanical reflexes resulting from an increase in elastic recoil and elastic load. The major cause of hypoxemia is ventilation and perfusion (V/Q) mismatching, not anatomic shunting or reduced oxygen diffusion as was previously suspected.62
During exercise, patients with IPF and may exhibit evidence of pulmonary hypertension, even in early cases that have preserved lung function at rest. Pulmonary hypertension can also be present at rest, and is an expected finding, once the vital capacity drops below 50% of predicted or the diffusing capacity falls below 45% of predicted. The presence of pulmonary hypertension may be a predictor of poor outcome yet may not correlate with lung function.63
 RADIOLOGY
RADIOLOGY
Conventional Chest Radiograph
The chest radiograph is abnormal in nearly all patients with IPF (Fig. 56-1). Yet, in up to 10% of patients with histologically proven IPF, the chest film might be normal. In most of these cases, the use of HRCT will uncover evidence of the disease.20
The most common abnormalities seen on a conventional chest film are reticular opacities. In other words there is an appearance of net-like linear and curvilinear densities. Reticular markings may be found bilaterally, in an asymmetrical distribution with a predilection for the lower lobes. A course reticular pattern on the plain radiograph, taking the form of translucent “honeycombing,” will emerge late in the course of disease and portends a poor prognosis. The chest radiograph lacks specificity for the diagnosis of IPF. The correct diagnosis is made on the conventional radiograph in less than 50% of cases. In addition, the interpretation of conventional radiographs with an interstitial pattern shows poor interobserver agreement. Studies have examined this particular characteristic and report that concordance between radiologists is only 70%.64,65
High-Resolution Computed Tomography
Development of the high-resolution CT scanner has revolutionized the diagnostic evaluation of the ILDs. HRCT allows a detailed examination of the lung parenchyma by creating 1- to 2-mm thin slices of the chest. HRCT uses a computerized reconstruction algorithm to maximize spatial resolution. This generates much improved image clarity such that the specificity of interpretations is increased, interobserver variability is reduced, and the overall accuracy of diagnosis is enhanced. HRCT scanning allows for the earlier diagnosis of IPF and permits the identification of alternate patterns of disease. The primary role of HRCT in the diagnostic evaluation of ILD is the discrimination of typical IPF from the other ILDs. Given the utility and availability of scanners, HRCT has become the primary diagnostic tool for identifying IPF.
The HRCT appearance of IPF is characterized by patchy, predominantly peripheral, predominantly subpleural, and bibasilar reticular opacities (Fig. 56-2). Ground-glass opacities can be found, but should occupy no more than a limited amount of territory. Areas that are severely involved with reticular markings may also demonstrate traction bronchiectasis. The presence of subpleural honeycombing (small, round translucencies with a density equal to that of air), traction bronchiectasis, and thickened interlobular septae will increase the specificity of the CT scan for diagnosing IPF. Several studies have examined the diagnostic accuracy of HRCT scans in IPF.64–68 Studies were conducted in which observers were asked to determine a radiographic diagnosis that was then compared with the histopathology of UIP as the “gold standard.” In the hands of experienced observers, radiographic diagnosis of IPF has a reported specificity and positive predictive value for IPF histology that exceeds 90%.66–68 However, the “confident” HRCT is not a sensitive tool for the diagnosis of IPF.66 The full spectrum of a “confident” radiographic pattern will only be seen in two-thirds of biopsy-proven IPF. One-third of IPF cases will not show a definitive CT pattern and would be missed if the HRCT was relied upon exclusively (Fig. 56-3). In such cases, and in the right clinical context, an SLB should be considered to clarify the diagnosis. Nonetheless it has become apparent that, in the right clinical setting, an experienced radiologist can diagnose IPF by the HRCT with considerable accuracy, obviating the need for biopsy.
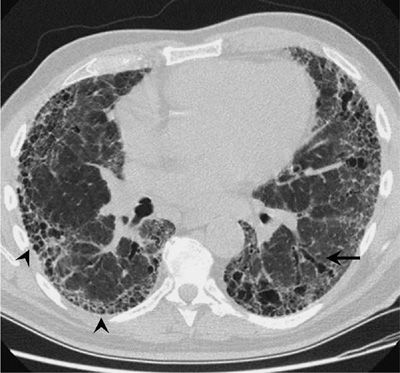
Figure 56-2 Computed tomography scan illustrates the “classic” features of idiopathic pulmonary fibrosis (IPF). Bilateral, peripheral, and subpleural reticular infiltrates are evident. The presence of advanced fibrosis is indicated by honeycomb changes (arrowheads) and traction bronchiectasis (arrow). These features permit experienced clinicians to make a confident radiographic diagnosis of IPF.
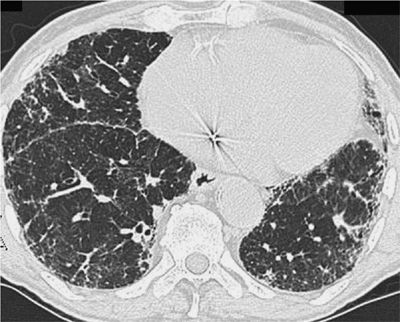
Figure 56-3 Computed tomography scan of an 81-year-old man with biopsy-proven idiopathic pulmonary fibrosis. A peripheral distribution of reticular opacities is demonstrated. Honeycombing and traction bronchiectasis are notably absent. In the absence of specific findings, a surgical lung biopsy was needed to make a diagnosis.
Given the evolving importance of HRCT in the diagnosis of IPF, CT scan criteria were defined during the most recent expert consensus statement on the diagnosis of IPF.20 HRCT patterns were separated into three groups: A UIP pattern; a possible UIP pattern; and a pattern labeled “inconsistent with UIP.” The UIP pattern has four features: (1) Subpleural, basal predominance of disease; (2) reticular abnormality; (3) honeycombing with or without traction bronchiectasis; and (4) absence of any inconsistent features. The inconsistent features include (1) upper or midlung predominance; (2) peribronchovascular predominance; (3) extensive ground-glass abnormalities that are greater than the amount of reticulation; (4) profuse micronodules; (5) multiple discrete cysts that are located away from areas of honeycombing; (6) diffuse mosaic attenuation/air trapping; and (7) consolidation in bronchopulmonary segments.20 Features defined as consistent with a “possible UIP pattern” were those of the UIP pattern except without evidence of honeycombing.
 BRONCHOALVEOLAR LAVAGE
BRONCHOALVEOLAR LAVAGE
An enormous amount of scientific information has been obtained by analyzing the content of BAL fluid from patients with IPF. Notable increases of immune cells (neutrophils, eosinophils, and activated alveolar macrophages) are present in BAL fluid from IPF. In addition, BAL has aided in the identification of cytokines, growth factors, and other cellular products that are now implicated in the pathogenesis of IPF. As a research tool, BAL has been immensely valuable. However, the role of BAL in the clinical diagnosis of IPF remains limited. Though much effort has been invested in evaluating the clinical utility of this modality, study results have been contradictory and generally disappointing.69 Most samples of BAL from IPF patients demonstrate simultaneous increases of several effector cell types including neutrophils (70%–90% of patients); eosinophils (40%–60% of patients); and lymphocytes (10%–20% of patients).20 Despite this finding, studies have failed to demonstrate a clear distinction amongst pulmonary diseases based upon the predominant type of cell in the BAL fluid.62 As a consequence of this, in standard practice, BAL is no longer generally recommended for the routine evaluation of IPF.
 PATHOLOGY
PATHOLOGY
Previously, SLB was recommended to confirm all cases of suspected IPF. With the advent and availability of HRCT this is no longer the case, as the positive predictive value of HRCT is comparable and in some studies better than that of biopsy.70 Despite this fact, SLB remains critical to the diagnosis of IPF in the context of HRCT scans that are equivocal (e.g., without the full complement of radiographic features that are expected in IPF). Biopsy may be achieved by either open thoracotomy or by video-assisted thoracoscopy (VATS). VATS is preferred as it has been associated with less morbidity and shorter hospital stays compared to open biopsy. An SLB provides the best sample from which to distinguish UIP from other forms of IIP. The recommendation is that the SLB be taken from at least two lobes, preferably the upper and lower lobes. The basis of this recommendation is from studies that identified that different pathologies could exist in different sections of the lung. Despite the coexisting patterns, if one of these had UIP pathology then the patient’s clinical course followed that of the UIP pathology.71 Transbronchial biopsies are less helpful in identifying IPF lesions because the small size of the sample prohibits the pathologist from identifying all the necessary features for a confident pathologic diagnosis of IPF.
The gross appearance of an IPF sample may be normal but often has a distinctive nodular pleural surface that has been likened to cirrhosis. The histopathologic lesion associated with IPF is UIP. This lesion is defined by a variegated structure. Normal lung alternates with patchy collagen fibrosis (Figs. 56-4 and 56-5). The fibrosis takes the form of alveolar septal thickening with a predominantly subpleural distribution. Whirls of fibroblasts embedded in a loose extracellular matrix embody the fibroblastic foci that are found in numerous quantities at the leading edge of dense scar (Figs. 56-4 and 56-5). Interstitial inflammation is present but remains scant and confined to areas of fibrosis. This limited inflammation consists of lymphocytes and plasma cells. Associated hyperplasia of the type 2 pneumocytes is found within areas of active inflammation. Areas that contain dense collagen may develop cystic structures, which may be filled with mucin or lined by bronchiolar epithelium. These cysts are referred to as microscopic honeycomb change. Hyaline membranes and organized alveolar exudates are absent. Occasionally alveolar macrophages are present.
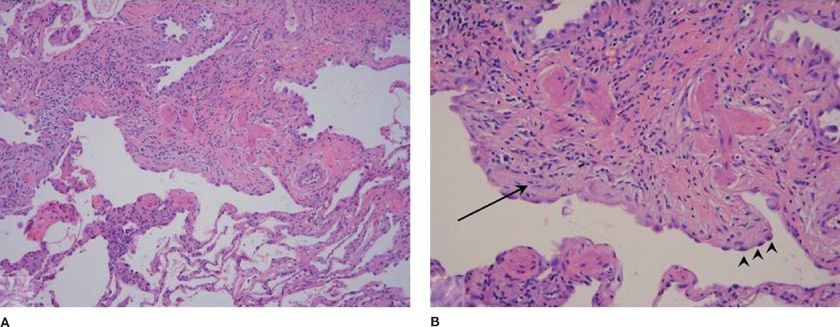
Figure 56-4 A. Low-magnification photomicrograph of usual interstitial pneumonia (UIP) showing the characteristic heterogeneous involvement of the parenchyma. Zones of interstitial fibrosis are seen alternating with areas of normal lung. B. Higher magnification demonstrates enlarged cystic air spaces lined with hyperplastic alveolar epithelium (arrowheads). Beneath the mucosal layer is an advancing region of young fibrosis containing loose extracellular matrix (pale pink staining) and fibroblasts (arrow).
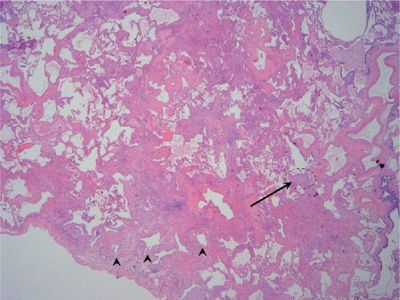
Figure 56-5 Scanning view of usual interstitial pneumonia (UIP) demonstrates the characteristic variegated appearance of UIP. Note the honeycomb change (arrowheads) present in the region of dense fibrosis adjacent to the pleural surface. A fibroblast focus (arrow) is seen at the leading edge of advancing fibrosis.
The UIP pathologic pattern exhibits a wide range of severity with regards to the extent of honeycomb change and the extent of involved lung. A history of smoking may alter the histopathologic appearance of UIP. Emphysematous change can be superimposed upon UIP. Pigmented alveolar macrophages, the hallmark feature of RB-ILD and DIP pathologic patterns, may be present in small number in UIP lesions from former or current smokers.
The UIP pattern can be found in other diseases besides IPF. The presence of granulomas in a UIP lesion favors a diagnosis of fibronodular sarcoidosis or chronic hypersensitivity pneumonitis. Asbestos bodies found within a UIP pattern suggest the diagnosis of asbestosis. The histopathologic pattern of UIP can also be found in several conditions other than IPF. UIP can be found in association with connective tissue diseases, asbestosis, chronic hypersensitivity pneumonitis, the Hermansky–Pudlak syndrome, neurofibromatosis, or in the setting of a toxic drug reaction (typically after administration of either bleomycin, methotrexate, nitrofurantoin, or amiodarone; [this is a partial list]). The identification of these conditions is largely a matter of correlation with the clinical history. It is important to note that the presence of honeycombing on biopsy is a nonspecific finding with a broad differential. Honeycombing is a common endpoint for a myriad of pathologic processes. Although honeycombing carries the connotation of end-stage fibrosis it can also occur in a focal distribution after any lung injury. Seen alone, honeycombing is not indicative of IPF.
To standardize the pathologic definition of UIP, a set of consensus criteria were established to represent expert opinion.20 The consensus definition allows for four pathologic categories: The UIP pattern; a probable UIP pattern; a possible UIP pattern; and a pattern referred to as “not UIP.” The UIP pattern requires (1) evidence of marked fibrosis/architectural distortion generally with honeycombing in a predominant subpleural distribution; (2) patchy involvement of the fibrosis in the lung parenchyma; (3) presence of fibroblastic foci; and (4) absence of features such as hyaline membranes, organizing pneumonia, granulomas, predominant airway-centered pathology, inflammatory cell infiltrate away from honeycombing or pathologic features suggestive of another disorder. The “probable” UIP pattern requires (1) evidence of marked fibrosis/architectural distortion with or without honeycombing; (2) absence of either patchy involvement or fibroblastic foci but not both; plus (3) an absence of features such as hyaline membranes, organizing pneumonia, granulomas, etc. In the right clinical context, “probable” UIP can be considered when honeycomb changes alone are present in the SLB. The “possible” UIP pattern incorporates (1) patchy or diffuse fibrosis within the pulmonary parenchyma; (2) in the absence of other criteria for a UIP pattern; and (3) in the absence of the features such as hyaline membranes, etc.
 DIAGNOSTIC ALGORITHM
DIAGNOSTIC ALGORITHM
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


