HYPOXIA AND CYANOSIS
HYPOXIA
The fundamental purpose of the cardiorespiratory system is to deliver O2 and nutrients to cells and to remove CO2 and other metabolic products from them. Proper maintenance of this function depends not only on intact cardiovascular and respiratory systems but also on an adequate number of red blood cells and hemoglobin and a supply of inspired gas containing adequate O2.
RESPONSES TO HYPOXIA
Decreased O2 availability to cells results in an inhibition of oxidative phosphorylation and increased anaerobic glycolysis. This switch from aerobic to anaerobic metabolism, the Pasteur effect, maintains some, albeit reduced, adenosine 5’-triphosphate (ATP) production. In severe hypoxia, when ATP production is inadequate to meet the energy requirements of ionic and osmotic equilibrium, cell membrane depolarization leads to uncontrolled Ca2+ influx and activation of Ca2+-dependent phospholipases and proteases. These events, in turn, cause cell swelling and, ultimately, cell death.
The adaptations to hypoxia are mediated, in part, by the upregulation of genes encoding a variety of proteins, including glycolytic enzymes such as phosphoglycerate kinase and phosphofructokinase, as well as the glucose transporters Glut-1 and Glut-2; and by growth factors, such as vascular endothelial growth factor (VEGF) and erythropoietin, which enhance erythrocyte production. The hypoxia-induced increase in expression of these key proteins is governed by the hypoxia-sensitive transcription factor, hypoxia-inducible factor-1 (HIF-1).
During hypoxia, systemic arterioles dilate, at least in part, by opening of KATP channels in vascular smooth-muscle cells due to the hypoxia-induced reduction in ATP concentration. By contrast, in pulmonary vascular smooth-muscle cells, inhibition of K+ channels causes depolarization which, in turn, activates voltage-gated Ca2+ channels raising the cytosolic [Ca2+] and causing smooth-muscle cell contraction. Hypoxia-induced pulmonary arterial constriction shunts blood away from poorly ventilated portions toward better ventilated portions of the lung; however, it also increases pulmonary vascular resistance and right ventricular afterload.
Effects on the central nervous system
Changes in the central nervous system (CNS), particularly the higher centers, are especially important consequences of hypoxia. Acute hypoxia causes impaired judgment, motor incoordination, and a clinical picture resembling acute alcohol intoxication. High-altitude illness is characterized by headache secondary to cerebral vasodilation, gastrointestinal symptoms, dizziness, insomnia, fatigue, or somnolence. Pulmonary arterial and sometimes venous constriction cause capillary leakage and high-altitude pulmonary edema (HAPE) (Chap. 5), which intensifies hypoxia, further promoting vasoconstriction. Rarely, high-altitude cerebral edema (HACE) develops, which is manifest by severe headache and papilledema and can cause coma. As hypoxia becomes more severe, the regulatory centers of the brainstem are affected, and death usually results from respiratory failure.
CAUSES OF HYPOXIA
Respiratory hypoxia
When hypoxia occurs from respiratory failure, PaO2 declines, and when respiratory failure is persistent, the hemoglobin-oxygen (Hb-O2) dissociation curve is displaced to the right, with greater quantities of O2released at any level of tissue PO2. Arterial hypoxemia, i.e., a reduction of O2 saturation of arterial blood (SaO2), and consequent cyanosis are likely to be more marked when such depression of PaO2 results from pulmonary disease than when the depression occurs as the result of a decline in the fraction of oxygen in inspired air (FIO2). In this latter situation, PacO2 falls secondary to anoxia-induced hyperventilation and the Hb-O2 dissociation curve is displaced to the left, limiting the decline in SaO2 at any level of PaO2.
The most common cause of respiratory hypoxia is ventilation-perfusion mismatch resulting from perfusion of poorly ventilated alveoli. Respiratory hypoxemia may also be caused by hypoventilation, in which case it is then associated with an elevation of PacO2. These two forms of respiratory hypoxia are usually correctable by inspiring 100% O2 for several minutes. A third cause of respiratory hypoxia is shunting of blood across the lung from the pulmonary arterial to the venous bed (intrapulmonary right-to-left shunting) by perfusion of nonventilated portions of the lung, as in pulmonary atelectasis or through pulmonary arteriovenous connections. The low PaO2 in this situation is only partially corrected by an FIO2 of 100%.
Hypoxia secondary to high altitude
As one ascends rapidly to 3000 m (~10,000 ft), the reduction of the O2 content of inspired air (FIO2) leads to a decrease in alveolar PO2 to approximately 60 mmHg, and a condition termed high-altitude illness develops (see earlier). At higher altitudes, arterial saturation declines rapidly and symptoms become more serious; and at 5000 m, unacclimated individuals usually cease to be able to function normally owing to the changes in CNS function described earlier.
Hypoxia secondary to right-to-left extrapulmonary shunting
From a physiologic viewpoint, this cause of hypoxia resembles intrapulmonary right-to-left shunting but is caused by congenital cardiac malformations, such as tetralogy of Fallot, transposition of the great arteries, and Eisenmenger’s syndrome (Chap. 19). As in pulmonary right-to-left shunting, the PaO2 cannot be restored to normal with inspiration of 100% O2.
Anemic hypoxia
A reduction in hemoglobin concentration of the blood is accompanied by a corresponding decline in the O2-carrying capacity of the blood. Although the PaO2 is normal in anemic hypoxia, the absolute quantity of O2 transported per unit volume of blood is diminished. As the anemic blood passes through the capillaries and the usual quantity of O2 is removed from it, the PO2 and saturation in the venous blood decline to a greater extent than normal.
Carbon monoxide (CO) intoxication
Hemoglobin that binds with CO (carboxyhemoglobin, COHb) is unavailable for O2 transport. In addition, the presence of COHb shifts the Hb-O2 dissociation curve to the left so that O2 is unloaded only at lower tensions, contributing further to tissue hypoxia.
Circulatory hypoxia
As in anemic hypoxia, the PaO2 is usually normal, but venous and tissue PO2 values are reduced as a consequence of reduced tissue perfusion and greater tissue O2 extraction. This pathophysiology leads to an increased arterial-mixed venous O2 difference (a-v-O2 difference), or gradient. Generalized circulatory hypoxia occurs in heart failure (Chap. 17) and in most forms of shock.
Specific organ hypoxia
Localized circulatory hypoxia may occur as a result of decreased perfusion secondary to arterial obstruction, as in localized atherosclerosis in any vascular bed, or as a consequence of vasoconstriction, as observed in Raynaud’s phenomenon (Chap. 39). Localized hypoxia may also result from venous obstruction and the resultant expansion of interstitial fluid causing arteriolar compression and, thereby, reduction of arterial inflow. Edema, which increases the distance through which O2 must diffuse before it reaches cells, can also cause localized hypoxia. In an attempt to maintain adequate perfusion to more vital organs in patients with reduced cardiac output secondary to heart failure or hypovolemic shock, vasoconstriction may reduce perfusion in the limbs and skin, causing hypoxia of these regions.
Increased O2 requirements
If the O2 consumption of tissues is elevated without a corresponding increase in perfusion, tissue hypoxia ensues and the PO2 in venous blood declines. Ordinarily, the clinical picture of patients with hypoxia due to an elevated metabolic rate, as in fever or thyrotoxicosis, is quite different from that in other types of hypoxia: the skin is warm and flushed owing to increased cutaneous blood flow that dissipates the excessive heat produced, and cyanosis is usually absent.
Exercise is a classic example of increased tissue O2 requirements. These increased demands are normally met by several mechanisms operating simultaneously: (1) increase in the cardiac output and ventilation and, thus, O2 delivery to the tissues; (2) a preferential shift in blood flow to the exercising muscles by changing vascular resistances in the circulatory beds of exercising tissues, directly and/or reflexly; (3) an increase in O2 extraction from the delivered blood and a widening of the arteriovenous O2 difference; and (4) a reduction in the pH of the tissues and capillary blood, shifting the Hb-O2 curve to the right, and unloading more O2 from hemoglobin. If the capacity of these mechanisms is exceeded, then hypoxia, especially of the exercising muscles, will result.
Improper oxygen utilization
Cyanide and several other similarly acting poisons cause cellular hypoxia. The tissues are unable to utilize O2, and, as a consequence, the venous blood tends to have a high O2 tension. This condition has been termed histotoxic hypoxia.
ADAPTATION TO HYPOXIA
An important component of the respiratory response to hypoxia originates in special chemosensitive cells in the carotid and aortic bodies and in the respiratory center in the brainstem. The stimulation of these cells by hypoxia increases ventilation, with a loss of CO2, and can lead to respiratory alkalosis. When combined with the metabolic acidosis resulting from the production of lactic acid, the serum bicarbonate level declines.
With the reduction of PaO2, cerebrovascular resistance decreases and cerebral blood flow increases in an attempt to maintain O2 delivery to the brain. However, when the reduction of PaO2 is accompanied by Hyperventilation and a reduction of PacO2, cerebrovascular resistance rises, cerebral blood flow falls, and tissue hypoxia intensifies.
The diffuse, systemic vasodilation that occurs in generalized hypoxia increases the cardiac output. In patients with underlying heart disease, the requirements of peripheral tissues for an increase of cardiac output with hypoxia may precipitate congestive heart failure. In patients with ischemic heart disease, a reduced PaO2 may intensify myocardial ischemia and further impair left ventricular function.
One of the important compensatory mechanisms for chronic hypoxia is an increase in the hemoglobin concentration and in the number of red blood cells in the circulating blood, i.e., the development of polycythemia secondary to erythropoietin production. In persons with chronic hypoxemia secondary to prolonged residence at a high altitude (>13,000 ft, 4200 m), a condition termed chronic mountain sickness develops. This disorder is characterized by a blunted respiratory drive, reduced ventilation, erythrocytosis, cyanosis, weakness, right ventricular enlargement secondary to pulmonary hypertension, and even stupor.
CYANOSIS
Cyanosis refers to a bluish color of the skin and mucous membranes resulting from an increased quantity of reduced hemoglobin (i.e., deoxygenated hemoglobin) or of hemoglobin derivatives (e.g., methemoglobin or sulfhemoglobin) in the small blood vessels of those tissues. It is usually most marked in the lips, nail beds, ears, and malar eminences. Cyanosis, especially if developed recently, is more commonly detected by a family member than the patient. The florid skin characteristic of polycythemia vera must be distinguished from the true cyanosis discussed here. A cherry-colored flush, rather than cyanosis, is caused by COHb.
The degree of cyanosis is modified by the color of the cutaneous pigment and the thickness of the skin, as well as by the state of the cutaneous capillaries. The accurate clinical detection of the presence and degree of cyanosis is difficult, as proved by oximetric studies. In some instances, central cyanosis can be detected reliably when the SaO2 has fallen to 85%; in others, particularly in dark-skinned persons, it may not be detected until it has declined to 75%. In the latter case, examination of the mucous membranes in the oral cavity and the conjunctivae rather than examination of the skin is more helpful in the detection of cyanosis.
The increase in the quantity of reduced hemoglobin in the mucocutaneous vessels that produces cyanosis may be brought about either by an increase in the quantity of venous blood as a result of dilation of the venules and venous ends of the capillaries or by a reduction in the SaO2 in the capillary blood. In general, cyanosis becomes apparent when the concentration of reduced hemoglobin in capillary blood exceeds 40 g/L (4 g/dL).
It is the absolute, rather than the relative, quantity of reduced hemoglobin that is important in producing cyanosis. Thus, in a patient with severe anemia, the relative quantity of reduced hemoglobin in the venous blood may be very large when considered in relation to the total quantity of hemoglobin in the blood. However, since the concentration of the latter is markedly reduced, the absolute quantity of reduced hemoglobin may still be small, and, therefore, patients with severe anemia and even marked arterial desaturation may not display cyanosis. Conversely, the higher the total hemoglobin content, the greater the tendency toward cyanosis; thus, patients with marked polycythemia tend to be cyanotic at higher levels of SaO2 than patients with normal hematocrit values. Likewise, local passive congestion, which causes an increase in the total quantity of reduced hemoglobin in the vessels in a given area, may cause cyanosis. Cyanosis is also observed when nonfunctional hemoglobin, such as methemoglobin or sulfhemoglobin, is present in blood.
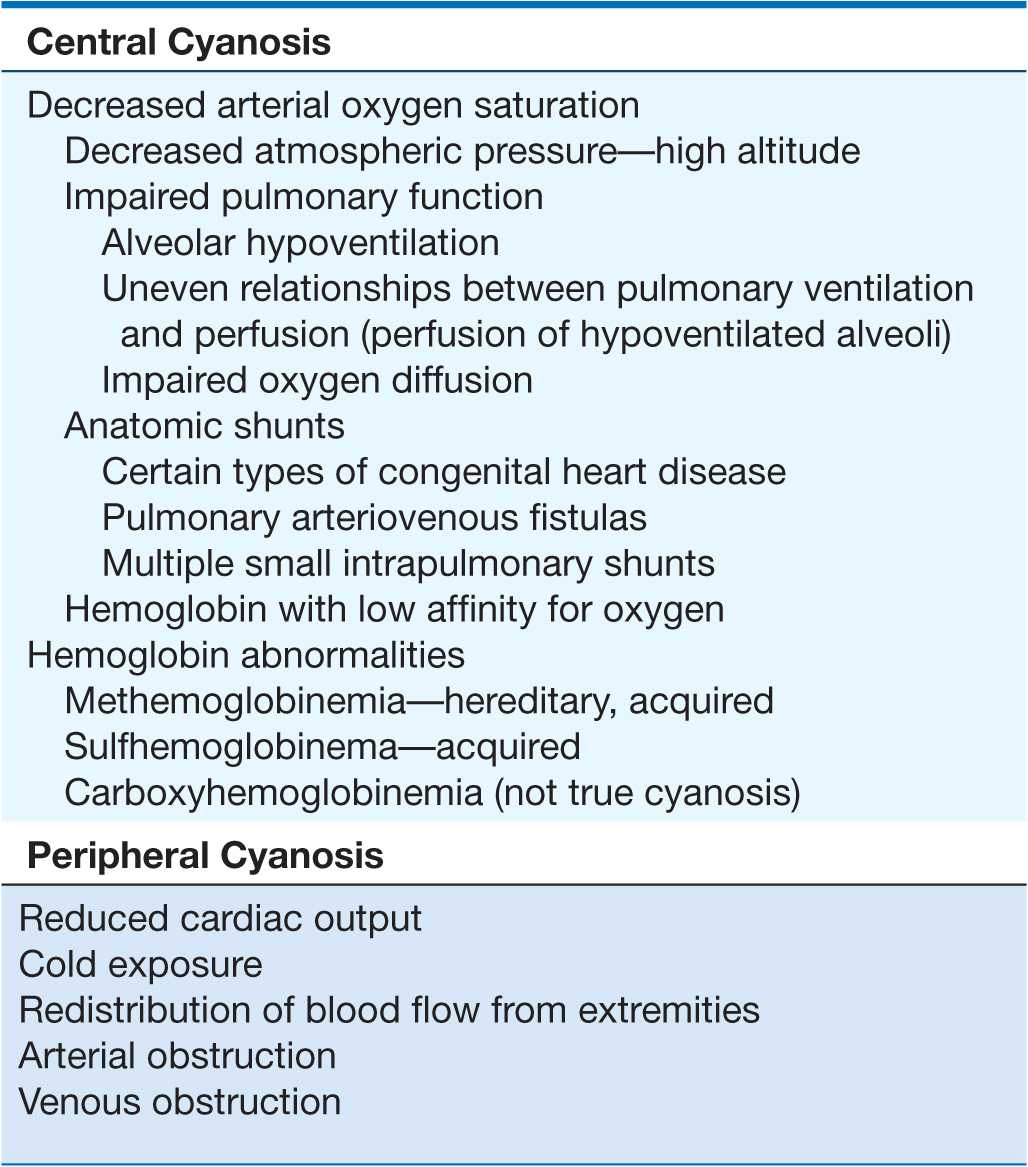
Cyanosis may be subdivided into central and peripheral types. In central cyanosis, the SaO2 is reduced or an abnormal hemoglobin derivative is present, and the mucous membranes and skin are both affected. Peripheral cyanosis is due to a slowing of blood flow and abnormally great extraction of O2 from normally saturated arterial blood; it results from vasoconstriction and diminished peripheral blood flow, such as occurs in cold exposure, shock, congestive failure, and peripheral vascular disease. Often in these conditions, the mucous membranes of the oral cavity or those beneath the tongue may be spared. Clinical differentiation between central and peripheral cyanosis may not always be simple, and in conditions such as cardiogenic shock with pulmonary edema there may be a mixture of both types.
DIFFERENTIAL DIAGNOSIS
Central cyanosis
(Table 6-1) Decreased SaO2 results from a marked reduction in the PaO2. This reduction may be brought about by a decline in the FIO2 without sufficient compensatory alveolar hyperventilation to maintain alveolar PO2. Cyanosis usually becomes manifest in an ascent to an altitude of 4000 m (13,000 ft).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


