HYPERTENSIVE VASCULAR DISEASE
Hypertension is one of the leading causes of the global burden of disease. Approximately 7.6 million deaths (13–15% of the total) and 92 million disability-adjusted life years worldwide were attributable to high blood pressure in 2001. Hypertension doubles the risk of cardiovascular diseases, including coronary heart disease (CHD), congestive heart failure (CHF), ischemic and hemorrhagic stroke, renal failure, and peripheral arterial disease. It often is associated with additional cardiovascular disease risk factors, and the risk of cardiovascular disease increases with the total burden of risk factors. Although antihypertensive therapy clearly reduces the risks of cardiovascular and renal disease, large segments of the hypertensive population are either untreated or inadequately treated.
EPIDEMIOLOGY
Blood pressure levels, the rate of age-related increases in blood pressure, and the prevalence of hypertension vary among countries and among subpopulations within a country. Hypertension is present in all populations except for a small number of individuals living in primitive, culturally isolated societies. In industrialized societies, blood pressure increases steadily during the first two decades of life. In children and adolescents, blood pressure is associated with growth and maturation. Blood pressure “tracks” over time in children and between adolescence and young adulthood. In the United States, average systolic blood pressure is higher for men than for women during early adulthood, although among older individuals the age-related rate of rise is steeper for women. Consequently, among individuals age 60 and older, systolic blood pressures of women are higher than those of men. Among adults, diastolic blood pressure also increases progressively with age until ~55 years, after which it tends to decrease. The consequence is a widening of pulse pressure (the difference between systolic and diastolic blood pressure) beyond age 60. The probability that a middle-aged or elderly individual will develop hypertension in his or her lifetime is 90%.
In the United States, based on results of the National Health and Nutrition Examination Survey (NHANES), approximately 30% (age-adjusted prevalence) of adults, or at least 65 million individuals, have hypertension (defined as any one of the following: systolic blood pressure ≥140 mmHg, diastolic blood pressure ≥90 mmHg, taking antihypertensive medications). Hypertension prevalence is 33.5% in non-Hispanic blacks, 28.9% in non-Hispanic whites, and 20.7% in Mexican Americans. The likelihood of hypertension increases with age, and among individuals age ≥60, the prevalence is 65.4%. Recent evidence suggests that the prevalence of hypertension in the United States may be increasing, possibly as a consequence of increasing obesity. The prevalence of hypertension and stroke mortality rates are higher in the southeastern United States than in other regions. In African Americans, hypertension appears earlier, is generally more severe, and results in higher rates of morbidity and mortality from stroke, left ventricular hypertrophy, CHF, and end-stage renal disease (ESRD) than in white Americans.
Both environmental and genetic factors may contribute to regional and racial variations in blood pressure and hypertension prevalence. Studies of societies undergoing “acculturation” and studies of migrants from a less to a more urbanized setting indicate a profound environmental contribution to blood pressure. Obesity and weight gain are strong, independent risk factors for hypertension. It has been estimated that 60% of hypertensives are >20% overweight. Among populations, hypertension prevalence is related to dietary NaCl intake, and the age-related increase in blood pressure may be augmented by a high NaCl intake. Low dietary intakes of calcium and potassium also may contribute to the risk of hypertension. The urine sodium-to-potassium ratio is a stronger correlate of blood pressure than is either sodium or potassium alone. Alcohol consumption, psychosocial stress, and low levels of physical activity also may contribute to hypertension.
Adoption, twin, and family studies document a significant heritable component to blood pressure levels and hypertension. Family studies controlling for a common environment indicate that blood pressure heritabilities are in the range 15–35%. In twin studies, heritability estimates of blood pressure are ~60% for males and 30–40% for females. High blood pressure before age 55 occurs 3.8 times more frequently among persons with a positive family history of hypertension.
GENETIC CONSIDERATIONS
![]() Although specific genetic variants have been identified in rare Mendelian forms of hypertension (Table 37-5), these variants are not applicable to the vast majority (>98%) of patients with essential hypertension. For most individuals, it is likely that hypertension represents a polygenic disorder in which a combination of genes acts in concert with environmental exposures to make only a modest contribution to blood pressure. Further, different subsets of genes may lead to different phenotypes associated with hypertension, e.g., obesity, dyslipidemia, insulin resistance.
Although specific genetic variants have been identified in rare Mendelian forms of hypertension (Table 37-5), these variants are not applicable to the vast majority (>98%) of patients with essential hypertension. For most individuals, it is likely that hypertension represents a polygenic disorder in which a combination of genes acts in concert with environmental exposures to make only a modest contribution to blood pressure. Further, different subsets of genes may lead to different phenotypes associated with hypertension, e.g., obesity, dyslipidemia, insulin resistance.
Several strategies are being utilized in the search for specific hypertension-related genes. Animal models (including selectively bred rats and congenic rat strains) provide a powerful approach for evaluating genetic loci and genes associated with hypertension. Comparative mapping strategies allow for the identification of syntenic genomic regions between the rat and human genomes that may be involved in blood pressure regulation. In association studies, different alleles (or combinations of alleles at different loci) of specific candidate genes or chromosomal regions are compared in hypertensive patients and normotensive control subjects. Current evidence suggests that genes that encode components of the renin-angiotensin-aldosterone system, along with angiotensinogen and angiotensin-converting enzyme (ACE) polymorphisms, may be related to hypertension and to blood pressure sensitivity to dietary NaCl. The alphaadducin gene is thought to be associated with increased renal tubular absorption of sodium, and variants of this gene may be associated with hypertension and salt sensitivity of blood pressure. Other genes possibly related to hypertension include genes encoding the AT1 receptor, aldosterone synthase, and the β2 adrenoreceptor. Genomewide association studies involve rapidly scanning markers across the entire genome to identify loci (not specific genes) associated with an observable trait (e.g., blood pressure) or a particular disease. This strategy has been facilitated by the availability of dense genotyping chips and the International HapMap. To date, the results of candidate gene studies often have not been replicated, and in contrast to several other polygenic disorders, genomewide association studies have had limited success in identifying genetic determinants of hypertension.
Preliminary evidence suggests that there may also be genetic determinants of target organ damage attributed to hypertension. Family studies indicate significant heritability of left ventricular mass, and there is considerable individual variation in the responses of the heart to hypertension. Family studies and variations in candidate genes associated with renal damage suggest that genetic factors also may contribute to hypertensive nephropathy. Specific genetic variants have been linked to CHD and stroke.
In the future, it is possible that DNA analysis will predict individual risk for hypertension and target organ damage and will identify responders to specific classes of antihypertensive agents. However, with the exception of the rare, monogenic hypertensive diseases, the genetic variants associated with hypertension remain to be confirmed, and the intermediate steps by which these variants affect blood pressure remain to be determined.
MECHANISMS OF HYPERTENSION
To provide a framework for understanding the pathogenesis of and treatment options for hypertensive disorders, it is useful to understand factors involved in the regulation of both normal and elevated arterial pressure. Cardiac output and peripheral resistance are the two determinants of arterial pressure (Fig. 37-1). Cardiac output is determined by stroke volume and heart rate; stroke volume is related to myocardial contractility and to the size of the vascular compartment. Peripheral resistance is determined by functional and anatomic changes in small arteries (lumen diameter 100–400 μm) and arterioles.
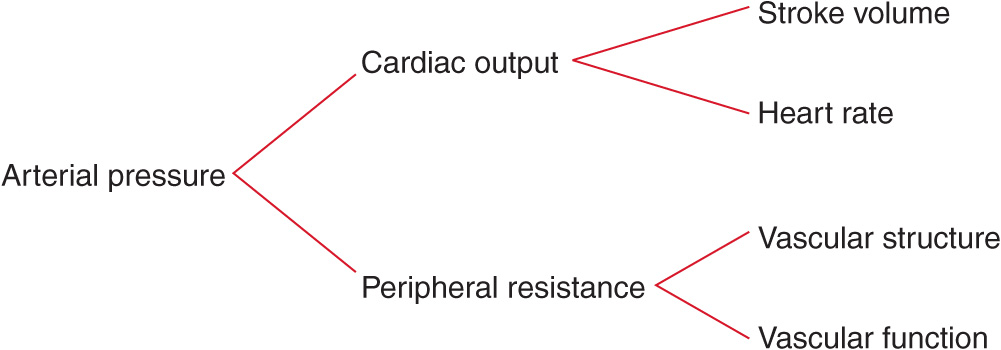
FIGURE 37-1
Determinants of arterial pressure.
INTRAVASCULAR VOLUME
Vascular volume is a primary determinant of arterial pressure over the long term. Sodium is predominantly an extracellular ion and is a primary determinant of the extracellular fluid volume. When NaCl intake exceeds the capacity of the kidney to excrete sodium, vascular volume initially expands and cardiac output increases. However, many vascular beds (including kidney and brain) have the capacity to autoregulate blood flow, and if constant blood flow is to be maintained in the face of increased arterial pressure, resistance within that bed must increase, since
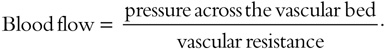
The initial elevation of blood pressure in response to vascular volume expansion may be related to an increase of cardiac output; however, over time, peripheral resistance increases and cardiac output reverts toward normal. The effect of sodium on blood pressure is related to the provision of sodium with chloride; nonchloride salts of sodium have little or no effect on blood pressure. As arterial pressure increases in response to a high NaCl intake, urinary sodium excretion increases and sodium balance is maintained at the expense of an increase in arterial pressure. The mechanism for this “pressure-natriuresis” phenomenon may involve a subtle increase in the glomerular filtration rate, decreased absorbing capacity of the renal tubules, and possibly hormonal factors such as atrial natriuretic factor. In individuals with an impaired capacity to excrete sodium, greater increases in arterial pressure are required to achieve natriuresis and sodium balance.
NaCl-dependent hypertension may be a consequence of a decreased capacity of the kidney to excrete sodium, due either to intrinsic renal disease or to increased production of a salt-retaining hormone (mineralocorticoid) resulting in increased renal tubular reabsorption of sodium. Renal tubular sodium reabsorption also may be augmented by increased neural activity to the kidney. In each of these situations, a higher arterial pressure may be required to achieve sodium balance. Conversely, salt-wasting disorders are associated with low blood pressure levels. ESRD is an extreme example of volume-dependent hypertension. In ~80% of these patients, vascular volume and hypertension can be controlled with adequate dialysis; in the other 20%, the mechanism of hypertension is related to increased activity of the renin-angiotensin system and is likely to be responsive to pharmacologic blockade of renin-angiotensin.
AUTONOMIC NERVOUS SYSTEM
The autonomic nervous system maintains cardiovascular homeostasis via pressure, volume, and chemoreceptor signals. Adrenergic reflexes modulate blood pressure over the short term, and adrenergic function, in concert with hormonal and volume-related factors, contributes to the long-term regulation of arterial pressure. The three endogenous catecholamines are norepinephrine, epinephrine, and dopamine. All three play important roles in tonic and phasic cardiovascular regulation.
The activities of the adrenergic receptors are mediated by guanosine nucleotide-binding regulatory proteins (G proteins) and by intracellular concentrations of downstream second messengers. In addition to receptor affinity and density, physiologic responsiveness to catecholamines may be altered by the efficiency of receptor-effector coupling at a site “distal” to receptor binding. The receptor sites are relatively specific both for the transmitter substance and for the response that occupancy of the receptor site elicits. Norepinephrine and epinephrine are agonists for all adrenergic receptor subtypes, although with varying affinities. Based on their physiology and pharmacology, adrenergic receptors have been divided into two principal types: α and β. These types have been differentiated further into α.1, α2, β1, and β,2 receptors. Recent molecular cloning studies have identified several additional subtypes. α Receptors are occupied and activated more avidly by norepinephrine than by epinephrine, and the reverse is true for β receptors. α1 Receptors are located on postsynaptic cells in smooth muscle and elicit vasoconstriction. α2 Receptors are localized on presynaptic membranes of postganglionic nerve terminals that synthesize norepinephrine. When activated by catecholamines, α2 receptors act as negative feedback controllers, inhibiting further norepinephrine release. In the kidney, activation of α1-adrenergic receptors increases renal tubular reabsorption of sodium. Different classes of antihypertensive agents either inhibit α1 receptors or act as agonists of α2 receptors and reduce systemic sympathetic outflow. Activation of myocardial β1 receptors stimulates the rate and strength of cardiac contraction and consequently increases cardiac output. β1 Receptor activation also stimulates renin release from the kidney. Another class of antihypertensive agents acts by inhibiting β1 receptors. Activation of α2 receptors by epinephrine relaxes vascular smooth muscle and results in vasodilation.
Circulating catecholamine concentrations may affect the number of adrenoreceptors in various tissues. Downregulation of receptors may be a consequence of sustained high levels of catecholamines and provides an explanation for decreasing responsiveness, or tachyphylaxis, to catecholamines. For example, orthostatic hypotension frequently is observed in patients with pheochromocytoma, possibly due to the lack of norepinephrine-induced vasoconstriction with assumption of the upright posture. Conversely, with chronic reduction of neurotransmitter substances, adrenoreceptors may increase in number or be upregulated, resulting in increased responsiveness to the neurotransmitter. Chronic administration of agents that block adrenergic receptors may result in upregulation, and withdrawal of those agents may produce a condition of temporary hypersensitivity to sympathetic stimuli. For example, clonidine is an antihypertensive agent that is a centrally acting α2 agonist that inhibits sympathetic outflow. Rebound hypertension may occur with the abrupt cessation of clonidine therapy, probably as a consequence of upregulation of α1 receptors.
Several reflexes modulate blood pressure on a minute-to-minute basis. One arterial baroreflex is mediated by stretch-sensitive sensory nerve endings in the carotid sinuses and the aortic arch. The rate of firing of these baroreceptors increases with arterial pressure, and the net effect is a decrease in sympathetic outflow, resulting in decreases in arterial pressure and heart rate. This is a primary mechanism for rapid buffering of acute fluctuations of arterial pressure that may occur during postural changes, behavioral or physiologic stress, and changes in blood volume. However, the activity of the baroreflex declines or adapts to sustained increases in arterial pressure such that the baroreceptors are reset to higher pressures. Patients with autonomic neuropathy and impaired baroreflex function may have extremely labile blood pressures with difficult-to-control episodic blood pressure spikes associated with tachycardia.
In both normal-weight and obese individuals, hypertension often is associated with increased sympathetic outflow. Based on recordings of postganglionic muscle nerve activity (detected by a microelectrode inserted in a peroneal nerve in the leg), sympathetic outflow tends to be higher in hypertensive than in normotensive individuals. Sympathetic outflow is increased in obesity-related hypertension and in hypertension associated with obstructive sleep apnea. Baroreceptor activation via electrical stimulation of carotid sinus afferent nerves has been shown to lower blood pressure in patients with “resistant” hypertension. Drugs that block the sympathetic nervous system are potent antihypertensive agents, indicating that the sympathetic nervous system plays a permissive, although not necessarily a causative, role in the maintenance of increased arterial pressure.
Pheochromocytoma is the most blatant example of hypertension related to increased catecholamine production, in this instance by a tumor. Blood pressure can be reduced by surgical excision of the tumor or by pharmacologic treatment with an α1 receptor antagonist or with an inhibitor of tyrosine hydroxylase, the rate-limiting step in catecholamine biosynthesis.
RENIN-ANGIOTENSIN-ALDOSTERONE
The renin-angiotensin-aldosterone system contributes to the regulation of arterial pressure primarily via the vasoconstrictor properties of angiotensin II and the sodium-retaining properties of aldosterone. Renin is an aspartyl protease that is synthesized as an enzymatically inactive precursor, prorenin. Most renin in the circulation is synthesized in the renal afferent renal arteriole. Prorenin may be secreted directly into the circulation or may be activated within secretory cells and released as active renin. Although human plasma contains two to five times more prorenin than renin, there is no evidence that prorenin contributes to the physiologic activity of this system. There are three primary stimuli for renin secretion: (1) decreased NaCl transport in the distal portion of the thick ascending limb of the loop of Henle that abuts the corresponding afferent arteriole (macula densa), (2) decreased pressure or stretch within the renal afferent arteriole (baroreceptor mechanism), and (3) sympathetic nervous system stimulation of renin-secreting cells via β1 adrenoreceptors. Conversely, renin secretion is inhibited by increased NaCl transport in the thick ascending limb of the loop of Henle, by increased stretch within the renal afferent arteriole, and by β1 receptor blockade. In addition, angiotensin II directly inhibits renin secretion due to angiotensin II type 1 receptors on juxtaglomerular cells, and renin secretion increases in response to pharmacologic blockade of either ACE or angiotensin II receptors.
Once released into the circulation, active renin cleaves a substrate, angiotensinogen, to form an inactive decapeptide, angiotensin I (Fig. 37-2). A converting enzyme, located primarily but not exclusively in the pulmonary circulation, converts angiotensin I to the active octapeptide, angiotensin II, by releasing the C-terminal histidyl-leucine dipeptide. The same converting enzyme cleaves a number of other peptides, including and thereby inactivating the vasodilator bradykinin. Acting primarily through angiotensin II type 1 (AT1) receptors on cell membranes, angiotensin II is a potent pressor substance, the primary tropic factor for the secretion of aldosterone by the adrenal zona glomerulosa, and a potent mitogen that stimulates vascular smooth-muscle cell and myocyte growth. Independent of its hemodynamic effects, angiotensin II may play a role in the pathogenesis of atherosclerosis through a direct cellular action on the vessel wall. An angiotensin II type 2 (AT2) receptor has been characterized. It is widely distributed in the kidney and has the opposite functional effects of the AT1 receptor. The AT2 receptor induces vasodilation, sodium excretion, and inhibition of cell growth and matrix formation. Experimental evidence suggests that the AT2 receptor improves vascular remodeling by stimulating smooth-muscle cell apoptosis and contributes to the regulation of glomerular filtration rate. AT1 receptor blockade induces an increase in AT2 receptor activity.
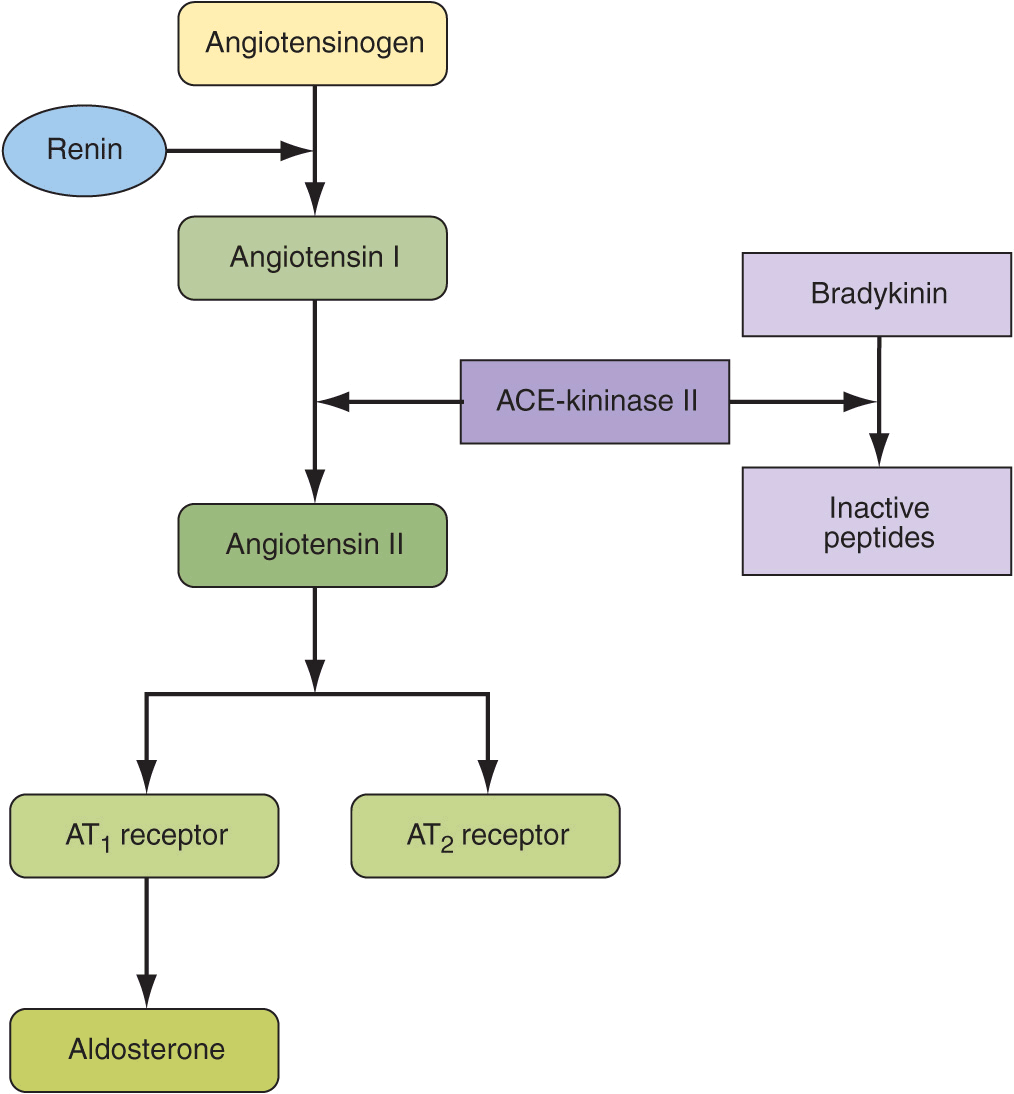
FIGURE 37-2
Renin-angiotensin-aldosterone axis.
Renin-secreting tumors are clear examples of renin-dependent hypertension. In the kidney, these tumors include benign hemangiopericytomas of the juxtaglomerular apparatus and, infrequently, renal carcinomas, including Wilms’ tumors. Renin-producing carcinomas also have been described in the lung, liver, pancreas, colon, and adrenals. In these instances, in addition to excision and/or ablation of the tumor, treatment of hypertension includes pharmacologic therapies targeted to inhibit angiotensin II production or action. Reno-vascular hypertension is another renin-mediated form of hypertension. Obstruction of the renal artery leads to decreased renal perfusion pressure, thereby stimulating renin secretion. Over time, as a consequence of secondary renal damage, this form of hypertension may become less renin dependent.
Angiotensinogen, renin, and angiotensin II are also synthesized locally in many tissues, including the brain, pituitary, aorta, arteries, heart, adrenal glands, kidneys, adipocytes, leukocytes, ovaries, testes, uterus, spleen, and skin. Angiotensin II in tissues may be formed by the enzymatic activity of renin or by other proteases, e.g., tonin, chymase, and cathepsins. In addition to regulating local blood flow, tissue angiotensin II is a mitogen that stimulates growth and contributes to modeling and repair. Excess tissue angiotensin II may contribute to atherosclerosis, cardiac hypertrophy, and renal failure and consequently may be a target for pharmacologic therapy to prevent target organ damage.
Angiotensin II is the primary tropic factor regulating the synthesis and secretion of aldosterone by the zona glomerulosa of the adrenal cortex. Aldosterone synthesis is also dependent on potassium, and aldosterone secretion may be decreased in potassium-depleted individuals. Although acute elevations of adrenocorticotropic hormone (ACTH) levels also increase aldosterone secretion, ACTH is not an important tropic factor for the chronic regulation of aldosterone.
Aldosterone is a potent mineralocorticoid that increases sodium reabsorption by amiloride-sensitive epithelial sodium channels (ENaC) on the apical surface of the principal cells of the renal cortical collecting duct. Electric neutrality is maintained by exchanging sodium for potassium and hydrogen ions. Consequently, increased aldosterone secretion may result in hypokalemia and alkalosis. Because potassium depletion may inhibit aldosterone synthesis, clinically, hypokalemia should be corrected before a patient is evaluated for hyperaldosteronism.
Mineralocorticoid receptors also are expressed in the colon, salivary glands, and sweat glands. Cortisol also binds to these receptors but normally functions as a less potent mineralocorticoid than aldosterone because cortisol is converted to cortisone by the enzyme 11β-hydroxysteroid dehydrogenase type 2. Cortisone has no affinity for the mineralocorticoid receptor. Primary aldosteronism is a compelling example of mineralocorticoid-mediated hypertension. In this disorder, adrenal aldosterone synthesis and release are independent of renin-angiotensin, and renin release is suppressed by the resulting volume expansion.
Aldosterone also has effects on nonepithelial targets. Aldosterone and/or mineralocorticoid receptor activation induces structural and functional alterations in the heart, kidney, and blood vessels, leading to myocardial fibrosis, nephrosclerosis, and vascular inflammation and remodeling, perhaps as a consequence of oxidative stress. These effects are amplified by a high salt intake. In animal models, high circulating aldosterone levels stimulate cardiac fibrosis and left ventricular hypertrophy, and spironolactone (an aldosterone antagonist) prevents aldosterone-induced myocardial fibrosis. Pathologic patterns of left ventricular geometry also have been associated with elevations of plasma aldosterone concentration in patients with essential hypertension as well as in patients with primary aldosteronism. In patients with CHF, low-dose spironolactone reduces the risk of progressive heart failure and sudden death from cardiac causes by 30%. Owing to a renal hemodynamic effect, in patients with primary aldosteronism, high circulating levels of aldosterone also may cause glomerular hyper-filtration and albuminuria. These renal effects are reversible after removal of the effects of excess aldosterone by adrenalectomy or spironolactone.
Increased activity of the renin-angiotensin-aldosterone axis is not invariably associated with hypertension. In response to a low-NaCl diet or to volume contraction, arterial pressure and volume homeostasis may be maintained by increased activity of the renin-angiotensinaldosterone axis. Secondary aldosteronism (i.e., increased aldosterone secondary to increased renin-angiotensin), but not hypertension, also is observed in edematous states such as CHF and liver disease.
VASCULAR MECHANISMS
Vascular radius and compliance of resistance arteries are also important determinants of arterial pressure. Resistance to flow varies inversely with the fourth power of the radius, and consequently, small decreases in lumen size significantly increase resistance. In hypertensive patients, structural, mechanical, or functional changes may reduce the lumen diameter of small arteries and arterioles. Remodeling refers to geometric alterations in the vessel wall without a change in vessel volume. Hypertrophic (increased cell size, and increased deposition of intercellular matrix) or eutrophic vascular remodeling results in decreased lumen size and hence contributes to increased peripheral resistance. Apoptosis, low-grade inflammation, and vascular fibrosis also contribute to remodeling. Lumen diameter also is related to elasticity of the vessel. Vessels with a high degree of elasticity can accommodate an increase of volume with relatively little change in pressure, whereas in a semirigid vascular system, a small increment in volume induces a relatively large increment of pressure.
Hypertensive patients have stiffer arteries, and arteriosclerotic patients may have particularly high systolic blood pressures and wide pulse pressures as a consequence of decreased vascular compliance due to structural changes in the vascular wall. Recent evidence suggests that arterial stiffness has independent predictive value for cardiovascular events. Clinically, a number of devices are available to evaluate arterial stiffness or compliance, including ultrasound and magnetic resonance imaging (MRI).
Ion transport by vascular smooth-muscle cells may contribute to hypertension-associated abnormalities of vascular tone and vascular growth, both of which are modulated by intracellular pH (pHi). Three ion transport mechanisms participate in the regulation of pHi: (1) Na+-H+ exchange, (2) Na+-dependent HCO3–-Cl–exchange, and (3) cation-independent HCO3–-Cl– exchange. Based on measurements in cell types that are more accessible than vascular smooth muscle (e.g., leukocytes, erythrocytes, platelets, skeletal muscle), activity of the Na+-H+ exchanger is increased in hypertension, and this may result in increased vascular tone by two mechanisms. First, increased sodium entry may lead to increased vascular tone by activating Na+-Ca2+ exchange and thereby increasing intracellular calcium. Second, increased pHi enhances calcium sensitivity of the contractile apparatus, leading to an increase in contractility for a given intracellular calcium concentration. Additionally, increased Na+-H+ exchange may stimulate growth of vascular smooth muscle cells by enhancing sensitivity to mitogens.
Vascular endothelial function also modulates vascular tone. The vascular endothelium synthesizes and releases a spectrum of vasoactive substances, including nitric oxide, a potent vasodilator. Endothelium-dependent vasodilation is impaired in hypertensive patients. This impairment often is assessed with high-resolution ultrasonography before and after the hyperemic phase of reperfusion that follows 5 min of forearm ischemia. Alternatively, endothelium-dependent vasodilation may be assessed in response to an intraarterially infused endothelium-dependent vasodilator, e.g., acetylcholine. Endothelin is a vasoconstrictor peptide produced by the endothelium, and orally active endothelin antagonists may lower blood pressure in patients with resistant hypertension.
Currently, it is not known if the hypertension-related vascular abnormalities of ion transport and endothelial function are primary alterations or secondary consequences of elevated arterial pressure. Limited evidence suggests that vascular compliance and endothelium-dependent vasodilation may be improved by aerobic exercise, weight loss, and anti-hypertensive agents. It remains to be determined whether these interventions affect arterial structure and stiffness via a blood pressure–independent mechanism and whether different classes of antihypertensive agents preferentially affect vascular structure and function.
PATHOLOGIC CONSEQUENCES OF HYPERTENSION
Hypertension is an independent predisposing factor for heart failure, coronary artery disease, stroke, renal disease, and peripheral arterial disease (PAD).
HEART
Heart disease is the most common cause of death in hypertensive patients. Hypertensive heart disease is the result of structural and functional adaptations leading to left ventricular hypertrophy, CHF, abnormalities of blood flow due to atherosclerotic coronary artery disease and microvascular disease, and cardiac arrhythmias.
Both genetic and hemodynamic factors contribute to left ventricular hypertrophy. Clinically, left ventricular hypertrophy can be diagnosed by electrocardiography, although echocardiography provides a more sensitive measure of left ventricular wall thickness. Individuals with left ventricular hypertrophy are at increased risk for CHD, stroke, CHF, and sudden death. Aggressive control of hypertension can regress or reverse left ventricular hypertrophy and reduce the risk of cardiovascular disease. It is not clear whether different classes of antihypertensive agents have an added impact on reducing left ventricular mass, independent of their blood pressure–lowering effect.
CHF may be related to systolic dysfunction, diastolic dysfunction, or a combination of the two. Abnormalities of diastolic function that range from asymptomatic heart disease to overt heart failure are common in hypertensive patients. Patients with diastolic heart failure have a preserved ejection fraction, which is a measure of systolic function. Approximately one-third of patients with CHF have normal systolic function but abnormal diastolic function. Diastolic dysfunction is an early consequence of hypertension-related heart disease and is exacerbated by left ventricular hypertrophy and ischemia. Cardiac catheterization provides the most accurate assessment of diastolic function. Alternatively, diastolic function can be evaluated by several noninvasive methods, including echocardiography and radionuclide angiography.
BRAIN
Stroke is the second most frequent cause of death in the world; it accounts for 5 million deaths each year, with an additional 15 million persons having nonfatal strokes. Elevated blood pressure is the strongest risk factor for stroke. Approximately 85% of strokes are due to infarction, and the remainder are due to either intracerebral or subarachnoid hemorrhage. The incidence of stroke rises progressively with increasing blood pressure levels, particularly systolic blood pressure in individuals >65 years. Treatment of hypertension convincingly decreases the incidence of both ischemic and hemorrhagic strokes.
Hypertension also is associated with impaired cognition in an aging population, and longitudinal studies support an association between midlife hypertension and late-life cognitive decline. Hypertension-related cognitive impairment and dementia may be a consequence of a single infarct due to occlusion of a “strategic” larger vessel or multiple lacunar infarcts due to occlusive small vessel disease resulting in subcortical white matter ischemia. Several clinical trials suggest that antihypertensive therapy has a beneficial effect on cognitive function, although this remains an active area of investigation.
Cerebral blood flow remains unchanged over a wide range of arterial pressures (mean arterial pressure of 50–150 mmHg) through a process termed autoregulation of blood flow. In patients with the clinical syndrome of malignant hypertension, encephalopathy is related to failure of autoregulation of cerebral blood flow at the upper pressure limit, resulting in vasodilation and hyperperfusion. Signs and symptoms of hypertensive encephalopathy may include severe headache, nausea and vomiting (often of a projectile nature), focal neurologic signs, and alterations in mental status. Untreated, hypertensive encephalopathy may progress to stupor, coma, seizures, and death within hours. It is important to distinguish hypertensive encephalopathy from other neurologic syndromes that may be associated with hypertension, e.g., cerebral ischemia, hemorrhagic or thrombotic stroke, seizure disorder, mass lesions, pseudotumor cerebri, delirium tremens, meningitis, acute intermittent porphyria, traumatic or chemical injury to the brain, and uremic encephalopathy.
KIDNEY
The kidney is both a target and a cause of hypertension. Primary renal disease is the most common etiology of secondary hypertension. Mechanisms of kidney-related hypertension include a diminished capacity to excrete sodium, excessive renin secretion in relation to volume status, and sympathetic nervous system overactivity. Conversely, hypertension is a risk factor for renal injury and end-stage renal disease. The increased risk associated with high blood pressure is graded, continuous, and present throughout the distribution of blood pressure above optimal pressure. Renal risk appears to be more closely related to systolic than to diastolic blood pressure, and black men are at greater risk than white men for developing ESRD at every level of blood pressure. Proteinuria is a reliable marker of the severity of chronic kidney disease and is a predictor of its progression. Patients with high urine protein excretion (>3 g/24 h) have a more rapid rate of progression than do those with lower protein excretion rates.
Atherosclerotic, hypertension-related vascular lesions in the kidney primarily affect preglomerular arterioles, resulting in ischemic changes in the glomeruli and postglomerular structures. Glomerular injury also may be a consequence of direct damage to the glomerular capillaries due to glomerular hyperperfusion. Studies of hypertension-related renal damage, primarily in experimental animals, suggest that loss of autoregulation of renal blood flow at the afferent arteriole results in transmission of elevated pressures to an unprotected glomerulus with ensuing hyperfiltration, hypertrophy, and eventual focal segmental glomerular sclerosis. With progressive renal injury there is a loss of autoregulation of renal blood flow and glomerular filtration rate, resulting in a lower blood pressure threshold for renal damage and a steeper slope between blood pressure and renal damage. The result may be a vicious cycle of renal damage and nephron loss leading to more severe hypertension, glomerular hyperfiltration, and further renal damage. Glomerular pathology progresses to glomerulosclerosis, and eventually the renal tubules may also become ischemic and gradually atrophic. The renal lesion associated with malignant hypertension consists of fibrinoid necrosis of the afferent arterioles, sometimes extending into the glomerulus, and may result in focal necrosis of the glomerular tuft.
Clinically, macroalbuminuria (a random urine albumin/creatinine ratio >300 mg/g) or microalbuminuria (a random urine albumin/creatinine ratio 30–300 mg/g) are early markers of renal injury. These are also risk factors for renal disease progression and cardiovascular disease.
PERIPHERAL ARTERIES
In addition to contributing to the pathogenesis of hypertension, blood vessels may be a target organ for atherosclerotic disease secondary to long-standing elevated blood pressure. Hypertensive patients with arterial disease of the lower extremities are at increased risk for future cardiovascular disease. Although patients with stenotic lesions of the lower extremities may be asymptomatic, intermittent claudication is the classic symptom of PAD. This is characterized by aching pain in the calves or buttocks while walking that is relieved by rest. The ankle-brachial index is a useful approach for evaluating PAD and is defined as the ratio of noninvasively assessed ankle to brachial (arm) systolic blood pressure. An ankle-brachial index <0.90 is considered diagnostic of PAD and is associated with >50% stenosis in at least one major lower limb vessel. Several studies suggest that an ankle-brachial index <0.80 is associated with elevated blood pressure, particularly systolic blood pressure.
DEFINING HYPERTENSION
From an epidemiologic perspective, there is no obvious level of blood pressure that defines hypertension. In adults, there is a continuous, incremental risk of cardiovascular disease, stroke, and renal disease across levels of both systolic and diastolic blood pressure. The Multiple Risk Factor Intervention Trial (MRFIT), which included >350,000 male participants, demonstrated a continuous and graded influence of both systolic and diastolic blood pressure on CHD mortality, extending down to systolic blood pressures of 120 mmHg. Similarly, results of a meta-analysis involving almost 1 million participants indicate that ischemic heart disease mortality, stroke mortality, and mortality from other vascular causes are directly related to the height of the blood pressure, beginning at 115/75 mmHg, without evidence of a threshold. Cardiovascular disease risk doubles for every 20-mmHg increase in systolic and 10-mmHg increase in diastolic pressure. Among older individuals, systolic blood pressure and pulse pressure are more powerful predictors of cardiovascular disease than is diastolic blood pressure.
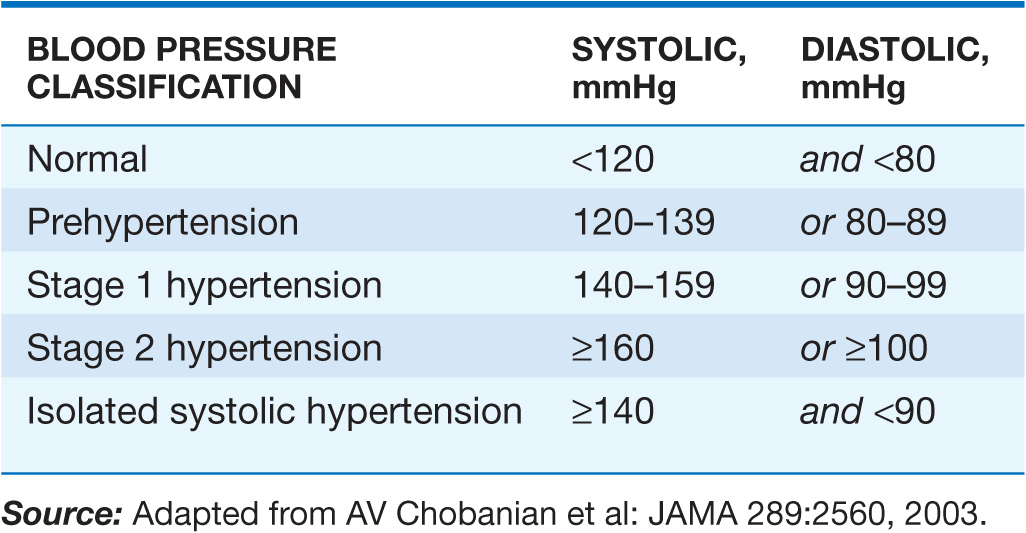
Clinically, hypertension may be defined as that level of blood pressure at which the institution of therapy reduces blood pressure–related morbidity and mortality. Current clinical criteria for defining hypertension generally are based on the average of two or more seated blood pressure readings during each of two or more outpatient visits. A recent classification recommends blood pressure criteria for defining normal blood pressure, prehypertension, hypertension (stages I and II), and isolated systolic hypertension, which is a common occurrence among the elderly (Table 37-1). In children and adolescents, hypertension generally is defined as systolic and/or diastolic blood pressure consistently >95th percentile for age, sex, and height. Blood pressures between the 90th and 95th percentiles are considered prehypertensive and are an indication for lifestyle interventions.
TABLE 37-1
BLOOD PRESSURE CLASSIFICATION
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


