HEART FAILURE AND COR PULMONALE
Douglas L. Mann ![]() Murali Chakinala
Murali Chakinala
HEART FAILURE
DEFINITION
Heart failure (HF) is a clinical syndrome that occurs in patients who, because of an inherited or acquired abnormality of cardiac structure and/or function, develop a constellation of clinical symptoms (dyspnea and fatigue) and signs (edema and rales) that lead to frequent hospitalizations, a poor quality of life, and a shortened life expectancy.
EPIDEMIOLOGY
 HF is a burgeoning problem worldwide, with more than 20 million people affected. The overall prevalence of HF in the adult population in developed countries is 2%. HF prevalence follows an exponential pattern, rising with age, and affects 6–10% of people over age 65. Although the relative incidence of HF is lower in women than in men, women constitute at least one-half the cases of HF because of their longer life expectancy. In North America and Europe, the lifetime risk of developing HF is approximately one in five for a 40-year-old. The overall prevalence of HF is thought to be increasing, in part because current therapies for cardiac disorders, such as myocardial infarction (MI), valvular heart disease, and arrhythmias, are allowing patients to survive longer. Very little is known about the prevalence or risk of developing HF in emerging nations because of the lack of population-based studies in those countries. Although HF once was thought to arise primarily in the setting of a depressed left ventricular (LV) ejection fraction (EF), epidemiologic studies have shown that approximately one-half of patients who develop HF have a normal or preserved EF (EF ≥40–50%). Accordingly, HF patients are now broadly categorized into one of two groups: (1) HF with a depressed EF (commonly referred to as systolic failure) or (2) HF with a preserved EF (commonly referred to as diastolic failure).
HF is a burgeoning problem worldwide, with more than 20 million people affected. The overall prevalence of HF in the adult population in developed countries is 2%. HF prevalence follows an exponential pattern, rising with age, and affects 6–10% of people over age 65. Although the relative incidence of HF is lower in women than in men, women constitute at least one-half the cases of HF because of their longer life expectancy. In North America and Europe, the lifetime risk of developing HF is approximately one in five for a 40-year-old. The overall prevalence of HF is thought to be increasing, in part because current therapies for cardiac disorders, such as myocardial infarction (MI), valvular heart disease, and arrhythmias, are allowing patients to survive longer. Very little is known about the prevalence or risk of developing HF in emerging nations because of the lack of population-based studies in those countries. Although HF once was thought to arise primarily in the setting of a depressed left ventricular (LV) ejection fraction (EF), epidemiologic studies have shown that approximately one-half of patients who develop HF have a normal or preserved EF (EF ≥40–50%). Accordingly, HF patients are now broadly categorized into one of two groups: (1) HF with a depressed EF (commonly referred to as systolic failure) or (2) HF with a preserved EF (commonly referred to as diastolic failure).
ETIOLOGY
As shown in Table 17-1, any condition that leads to an alteration in LV structure or function can predispose a patient to developing HF. Although the etiology of HF in patients with a preserved EF differs from that of patients with depressed EF, there is considerable overlap between the etiologies of these two conditions. In industrialized countries, coronary artery disease (CAD) has become the predominant cause in men and women and is responsible for 60–75% of cases of HF. Hypertension contributes to the development of HF in 75% of patients, including most patients with CAD. Both CAD and hypertension interact to augment the risk of HF, as does diabetes mellitus.
TABLE 17-1
ETIOLOGIES OF HEART FAILURE
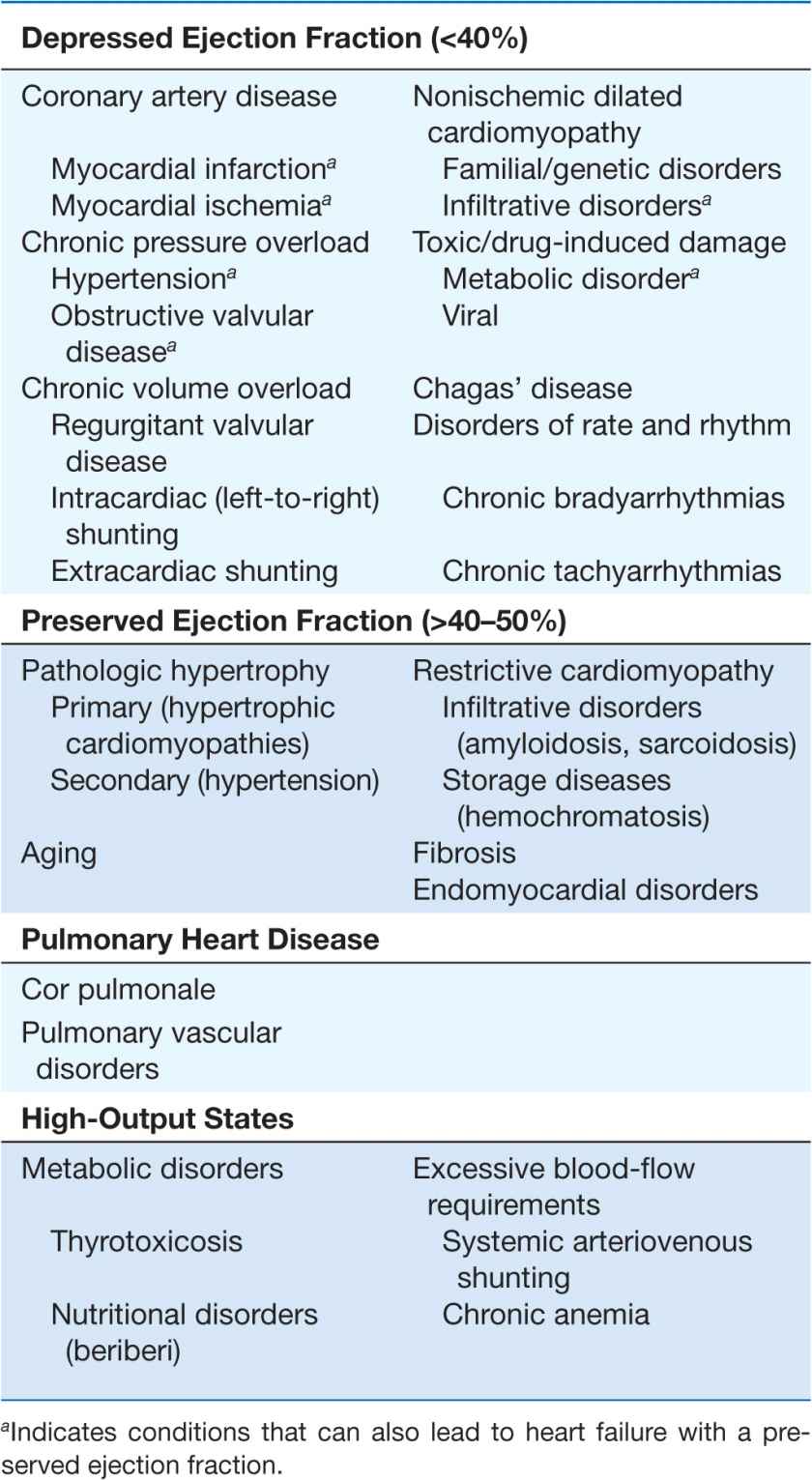
In 20–30% of the cases of HF with a depressed EF, the exact etiologic basis is not known. These patients are referred to as having nonischemic, dilated, or idiopathic cardiomyopathy if the cause is unknown (Chap. 21). Prior viral infection or toxin exposure (e.g., alcoholic or chemotherapeutic) also may lead to a dilated cardiomyopathy. Moreover, it is becoming increasingly clear that a large number of cases of dilated cardiomyopathy are secondary to specific genetic defects, most notably those in the cytoskeleton. Most forms of familial dilated cardiomyopathy are inherited in an autosomal dominant fashion. Mutations of genes that encode cytoskeletal proteins (desmin, cardiac myosin, vinculin) and nuclear membrane proteins (laminin) have been identified thus far. Dilated cardiomyopathy also is associated with Duchenne’s, Becker’s, and limb-girdle muscular dystrophies. Conditions that lead to a high cardiac output (e.g., arteriovenous fistula, anemia) are seldom responsible for the development of HF in a normal heart; however, in the presence of underlying structural heart disease, these conditions can lead to overt HF.
GLOBAL CONSIDERATIONS
 Rheumatic heart disease remains a major cause of HF in Africa and Asia, especially in the young. Hypertension is an important cause of HF in the African and African-American populations. Chagas’ disease is still a major cause of HF in South America. Not surprisingly, anemia is a frequent concomitant factor in HF in many developing nations. As developing nations undergo socioeconomic development, the epidemiology of HF is becoming similar to that of Western Europe and North America, with CAD emerging as the single most common cause of HF. Although the contribution of diabetes mellitus to HF is not well understood, diabetes accelerates atherosclerosis and often is associated with hypertension.
Rheumatic heart disease remains a major cause of HF in Africa and Asia, especially in the young. Hypertension is an important cause of HF in the African and African-American populations. Chagas’ disease is still a major cause of HF in South America. Not surprisingly, anemia is a frequent concomitant factor in HF in many developing nations. As developing nations undergo socioeconomic development, the epidemiology of HF is becoming similar to that of Western Europe and North America, with CAD emerging as the single most common cause of HF. Although the contribution of diabetes mellitus to HF is not well understood, diabetes accelerates atherosclerosis and often is associated with hypertension.
PROGNOSIS
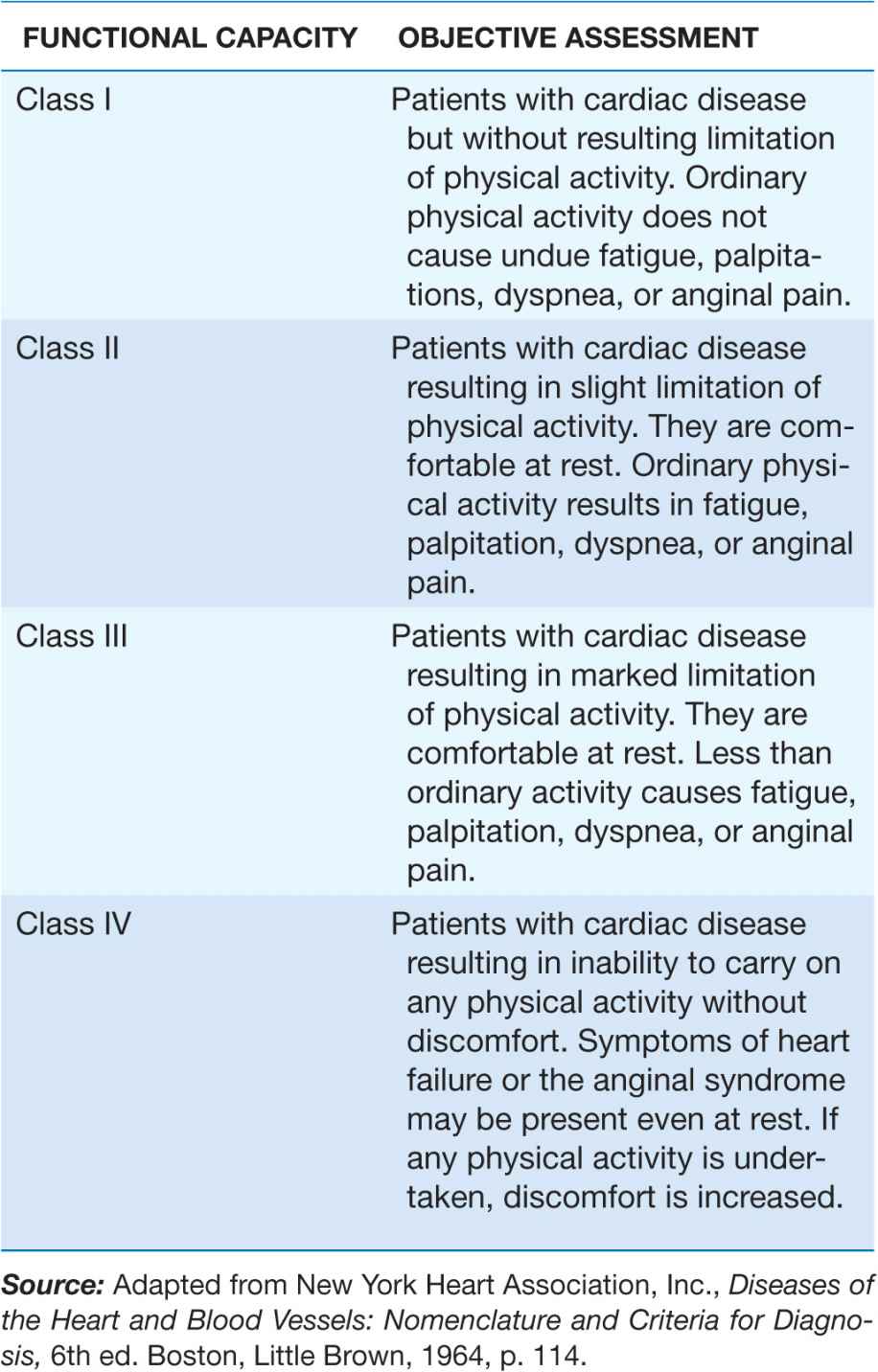
Despite many recent advances in the evaluation and management of HF, the development of symptomatic HF still carries a poor prognosis. Community-based studies indicate that 30–40% of patients die within 1 year of diagnosis and 60–70% die within 5 years, mainly from worsening HF or as a sudden event (probably because of a ventricular arrhythmia). Although it is difficult to predict prognosis in an individual, patients with symptoms at rest (New York Heart Association [NYHA] class IV) have a 30–70% annual mortality rate, whereas patients with symptoms with moderate activity (NYHA class II) have an annual mortality rate of 5–10%. Thus, functional status is an important predictor of patient outcome (Table 17-2).
TABLE 17-2
NEW YORK HEART ASSOCIATION CLASSIFICATION
PATHOGENESIS
Figure 17-1 provides a general conceptual framework for considering the development and progression of HF with a depressed EF. As shown, HF may be viewed as a progressive disorder that is initiated after an index event either damages the heart muscle, with a resultant loss of functioning cardiac myocytes, or, alternatively, disrupts the ability of the myocardium to generate force, thereby preventing the heart from contracting normally. This index event may have an abrupt onset, as in the case of a myocardial infarction (MI); it may have a gradual or insidious onset, as in the case of hemodynamic pressure or volume overloading; or it may be hereditary, as in the case of many of the genetic cardiomyopathies. Regardless of the nature of the inciting event, the feature that is common to each of these index events is that they all in some manner produce a decline in the pumping capacity of the heart. In most instances, patients remain asymptomatic or minimally symptomatic after the initial decline in pumping capacity of the heart or develop symptoms only after the dysfunction has been present for some time.
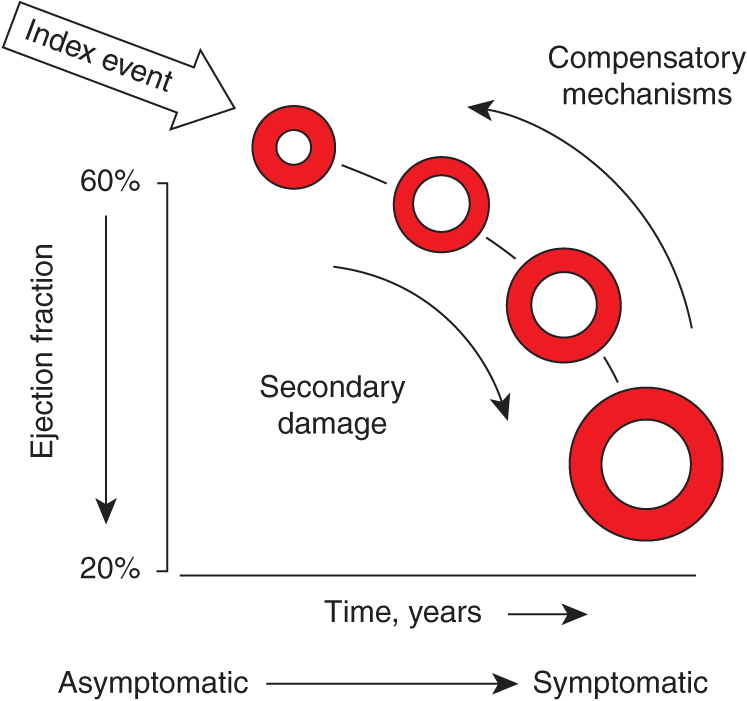
FIGURE 17-1
Pathogenesis of heart failure with a depressed ejection fraction. Heart failure begins after an index event produces an initial decline in the heart’s pumping capacity. After this initial decline in pumping capacity, a variety of compensatory mechanisms are activated, including the adrenergic nervous system, the renin-angiotensin-aldosterone system, and the cytokine system. In the short term, these systems are able to restore cardiovascular function to a normal homeostatic range with the result that the patient remains asymptomatic. However, with time the sustained activation of these systems can lead to secondary end-organ damage within the ventricle, with worsening left ventricular remodeling and subsequent cardiac decompensation. (From D Mann: Circulation 100:999, 1999.)
Although the precise reasons why patients with LV dysfunction may remain asymptomatic is not certain, one potential explanation is that a number of compensatory mechanisms become activated in the presence of cardiac injury and/or LV dysfunction allowing patients to sustain and modulate LV function for a period of months to years. The list of compensatory mechanisms that have been described thus far include (1) activation of the renin-angiotensin-aldosterone (RAA) and adrenergic nervous systems, which are responsible for maintaining cardiac output through increased retention of salt and water (Fig. 17-2), and (2) increased myocardial contractility. In addition, there is activation of a family of countervailing vasodilatory molecules, including the atrial and brain natriuretic peptides (ANP and BNP), prostaglandins (PGE2 and PGI2), and nitric oxide (NO), that offsets the excessive peripheral vascular vasoconstriction. Genetic background, sex, age, or environment may influence these compensatory mechanisms, which are able to modulate LV function within a physiologic/homeostatic range so that the functional capacity of the patient is preserved or is depressed only minimally. Thus, patients may remain asymptomatic or minimally symptomatic for a period of years; however, at some point patients become overtly symptomatic, with a resultant striking increase in morbidity and mortality rates. Although the exact mechanisms that are responsible for this transition are not known, as will be discussed later in this chapter, the transition to symptomatic HF is accompanied by increasing activation of neurohormonal, adrenergic, and cytokine systems that lead to a series of adaptive changes within the myocardium collectively referred to as LV remodeling.
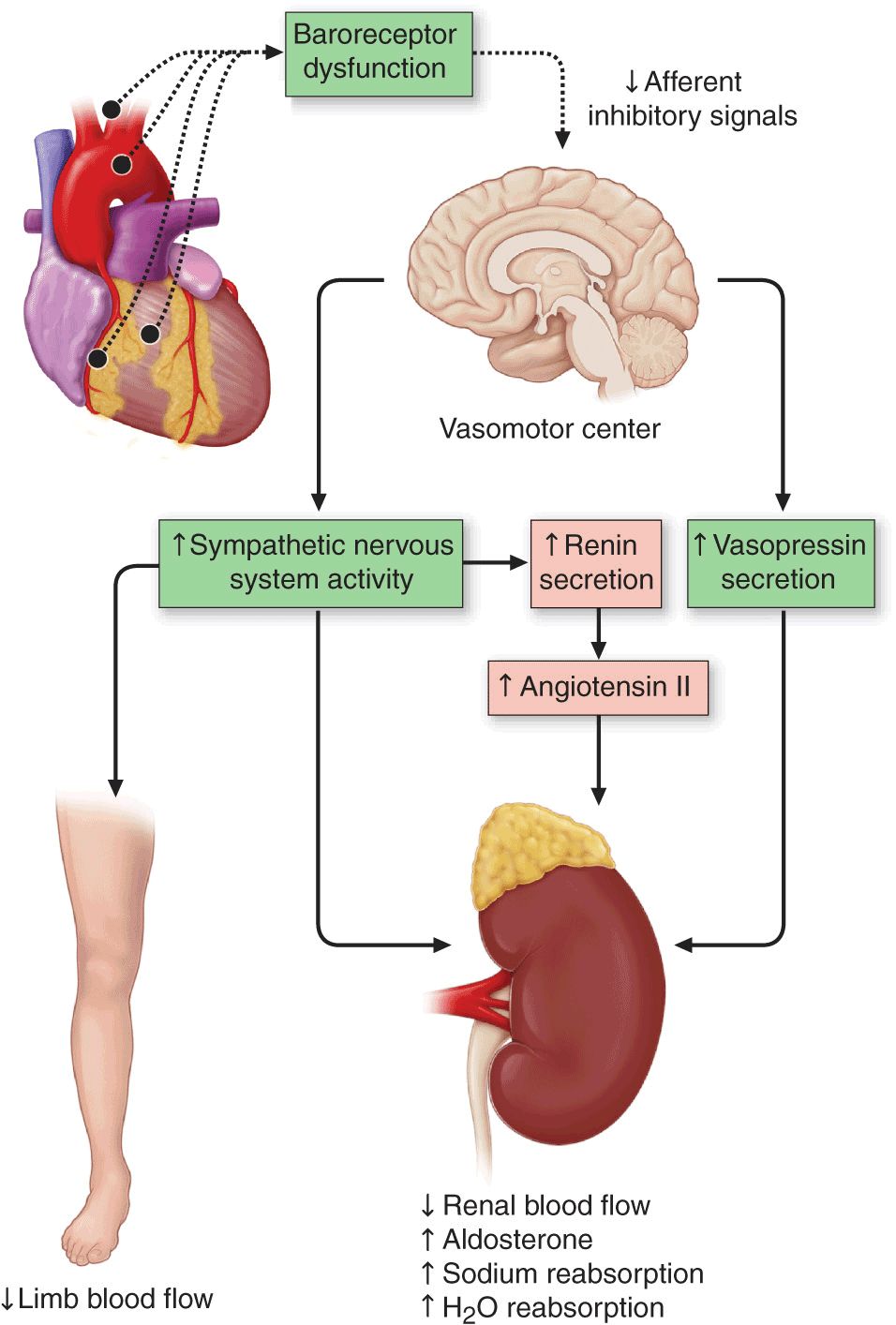
FIGURE 17-2
Activation of neurohormonal systems in heart failure. The decreased cardiac output in HF patients results in an “unloading” of high-pressure baroceptors (circles) in the left ventricle, carotid sinus, and aortic arch. This unloading of the peripheral baroreceptors leads to a loss of inhibitory parasympathetic tone to the central nervous system (CNS), with a resultant generalized increase in efferent sympathetic tone, and nonosmotic release of arginine vasopressin (AVP) from the pituitary. AVP (or antidiuretic hormone [ADH]) is a powerful vasoconstrictor that increases the permeability of the renal collecting ducts, leading to the reabsorption of free water. These afferent signals to the CNS also activate efferent sympathetic nervous system pathways that innervate the heart, kidney, peripheral vasculature, and skeletal muscles.
Sympathetic stimulation of the kidney leads to the release of renin, with a resultant increase in the circulating levels of angiotensin II and aldosterone. The activation of the reninangiotensin-aldosterone system promotes salt and water retention and leads to vasoconstriction of the peripheral vasculature, myocyte hypertrophy, myocyte cell death, and myocardial fibrosis. Although these neurohormonal mechanisms facilitate short-term adaptation by maintaining blood pressure, and hence perfusion to vital organs, the same neurohormonal mechanisms are believed to contribute to end-organ changes in the heart and the circulation and to the excessive salt and water retention in advanced HF. (Modified from A Nohria et al: Neurohormonal, renal and vascular adjustments, in Atlas of Heart Failure: Cardiac Function and Dysfunction, 4th ed, WS Colucci [ed]. Philadelphia, Current Medicine Group 2002, p. 104.)
In contrast to our understanding of the pathogenesis of HF with a depressed EF, our understanding of the mechanisms that contribute to the development of HF with a preserved EF is still evolving. That is, although diastolic dysfunction (see later in this chapter) was thought to be the only mechanism responsible for the development of HF with a preserved EF, community-based studies suggest that additional extracardiac mechanisms may be important, such as increased vascular stiffness and impaired renal function.
BASIC MECHANISMS OF HEART FAILURE
Systolic dysfunction
LV remodeling develops in response to a series of complex events that occur at the cellular and molecular levels (Table 17-3). These changes include (1) myocyte hypertrophy, (2) alterations in the contractile properties of the myocyte, (3) progressive loss of myocytes through necrosis, apoptosis, and autophagic cell death, (4) β-adrenergic desensitization, (5) abnormal myocardial energetics and metabolism, and (6) reorganization of the extracellular matrix with dissolution of the organized structural collagen weave surrounding myocytes and subsequent replacement by an interstitial collagen matrix that does not provide structural support to the myocytes. The biologic stimuli for these profound changes include mechanical stretch of the myocyte, circulating neurohormones (e.g., norepinephrine, angiotensin II), inflammatory cytokines (e.g., tumor necrosis factor [TNF]), other peptides and growth factors (e.g., endothelin), and reactive oxygen species (e.g., superoxide). The sustained overexpression of these biologically active molecules is believed to contribute to the progression of HF by virtue of the deleterious effects they exert on the heart and circulation. Indeed, this insight forms the clinical rationale for using pharmacologic agents that antagonize these systems (e.g., angiotensin-converting enzyme [ACE] inhibitors and beta blockers) in treating patients with HF.
TABLE 17-3
OVERVIEW OF LEFT VENTRICULAR REMODELING
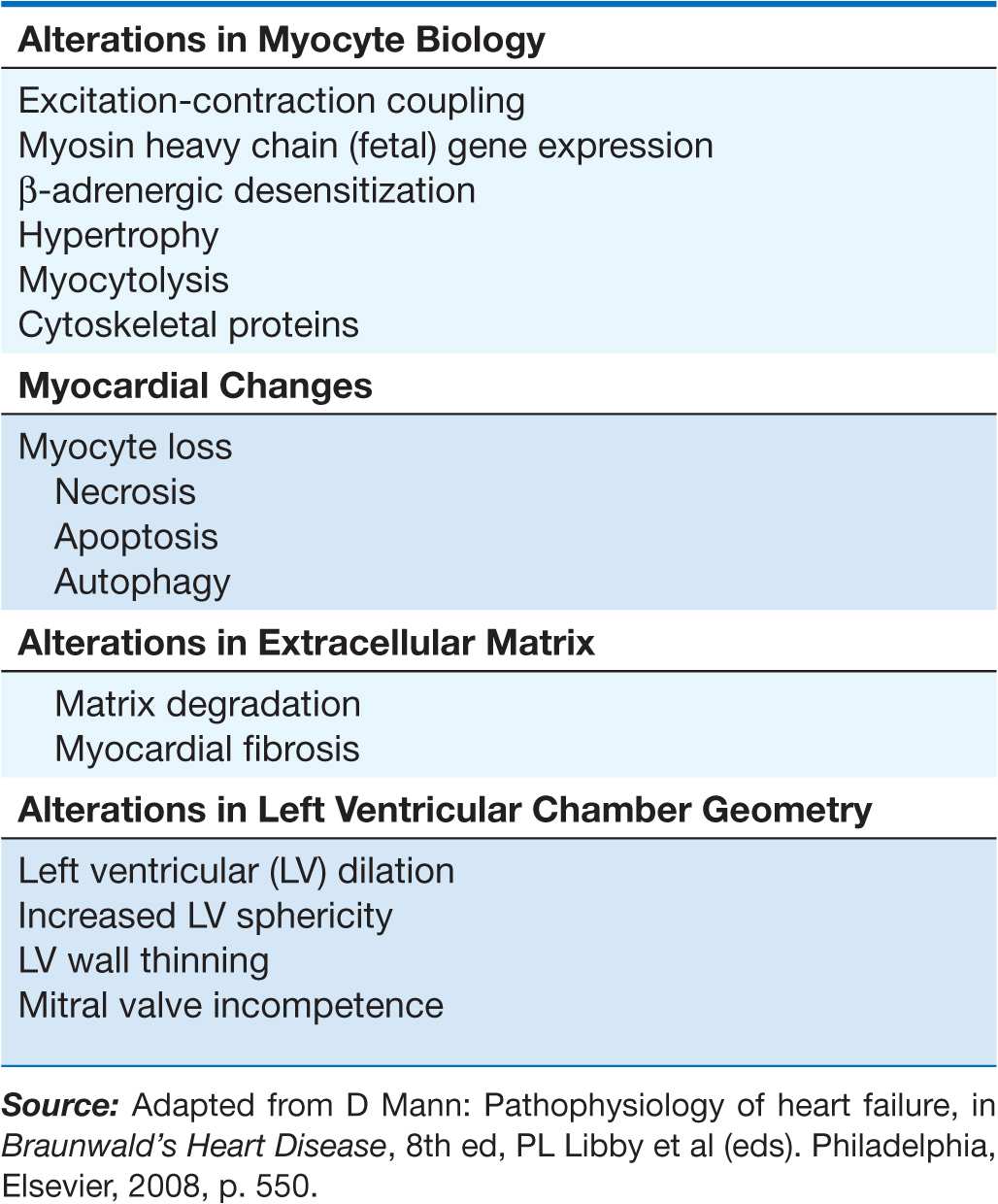
In order to understand how the changes that occur in the failing cardiac myocyte contribute to depressed LV systolic function in HF, it is instructive first to review the biology of the cardiac muscle cell (Chap. 1). Sustained neurohormonal activation and mechanical overload result in transcriptional and posttranscriptional changes in the genes and proteins that regulate excitation-contraction coupling and cross-bridge interaction (see Figs. 1-6 and 1-7). The changes that regulate excitation-contraction include decreased function of sarcoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA2A), resulting in decreased calcium uptake into the sarcoplasmic reticulum (SR), and hyperphosphorylation of the ryanodine receptor, leading to calcium leakage from the SR. The changes that occur in the cross-bridges include decreased expression of α-myosin heavy chain and increased expression of β-myosin heavy chain, myocytolysis, and disruption of the cytoskeletal links between the sarcomeres and the extracellular matrix. Collectively, these changes impair the ability of the myocyte to contract and therefore contribute to the depressed LV systolic function observed in patients with HF.
Diastolic dysfunction
Myocardial relaxation is an adenosine triphosphate (ATP)-dependent process that is regulated by uptake of cytoplasmic calcium into the SR by SERCA2A and extrusion of calcium by sarcolemmal pumps (see Fig. 1-7). Accordingly, reductions in ATP concentration, as occurs in ischemia, may interfere with these processes and lead to slowed myocardial relaxation. Alternatively, if LV filling is delayed because LV compliance is reduced (e.g., from hypertrophy or fibrosis), LV filling pressures will similarly remain elevated at end diastole (see Fig. 1-11). An increase in heart rate disproportionately shortens the time for diastolic filling, which may lead to elevated LV filling pressures, particularly in non-compliant ventricles. Elevated LV end-diastolic filling pressures result in increases in pulmonary capillary pressures, which can contribute to the dyspnea experienced by patients with diastolic dysfunction. In addition to impaired myocardial relaxation, increased myocardial stiffness secondary to cardiac hypertrophy and increased myocardial collagen content may contribute to diastolic failure. Importantly, diastolic dysfunction can occur alone or in combination with systolic dysfunction in patients with HF.
Left ventricular remodeling
Ventricular remodeling refers to the changes in LV mass, volume, and shape and the composition of the heart that occur after cardiac injury and/or abnormal hemodynamic loading conditions. LV remodeling may contribute independently to the progression of HF by virtue of the mechanical burdens that are engendered by the changes in the geometry of the remodeled LV. In addition to the increase in LV end-diastolic volume, LV wall thinning occurs as the left ventricle begins to dilate. The increase in wall thinning, along with the increase in afterload created by LV dilation, leads to a functional afterload mismatch that may contribute further to a decrease in stroke volume. Moreover, the high end-diastolic wall stress might be expected to lead to (1) hypoperfusion of the subendocardium, with resultant worsening of LV function, (2) increased oxidative stress, with the resultant activation of families of genes that are sensitive to free radical generation (e.g., TNF and interleukin 1β), and (3) sustained expression of stretch-activated genes (angiotensin II, endothelin, and TNF) and/or stretch activation of hypertrophic signaling pathways. Increasing LV dilation also results in tethering of the papillary muscles with resulting incompetence of the mitral valve apparatus and functional mitral regurgitation, which in turn leads to further hemodynamic overloading of the ventricle. Taken together, the mechanical burdens that are engendered by LV remodeling contribute to the progression of HF.
CLINICAL MANIFESTATIONS
Symptoms
The cardinal symptoms of HF are fatigue and shortness of breath. Although fatigue traditionally has been ascribed to the low cardiac output in HF, it is likely that skeletal-muscle abnormalities and other noncardiac comorbidities (e.g., anemia) also contribute to this symptom. In the early stages of HF, dyspnea is observed only during exertion; however, as the disease progresses, dyspnea occurs with less strenuous activity, and it ultimately may occur even at rest. The origin of dyspnea in HF is probably multifactorial (Chap. 5). The most important mechanism is pulmonary congestion with accumulation of interstitial or intraalveolar fluid, which activates juxtacapillary J receptors, which in turn stimulate the rapid, shallow breathing characteristic of cardiac dyspnea. Other factors that contribute to dyspnea on exertion include reductions in pulmonary compliance, increased airway resistance, respiratory muscle and/or diaphragm fatigue, and anemia. Dyspnea may become less frequent with the onset of right ventricular (RV) failure and tricuspid regurgitation.
 Orthopnea
Orthopnea
Orthopnea, which is defined as dyspnea occurring in the recumbent position, is usually a later manifestation of HF than is exertional dyspnea. It results from redistribution of fluid from the splanchnic circulation and lower extremities into the central circulation during recumbency, with a resultant increase in pulmonary capillary pressure. Nocturnal cough is a common manifestation of this process and a frequently overlooked symptom of HF. Orthopnea generally is relieved by sitting upright or sleeping with additional pillows. Although orthopnea is a relatively specific symptom of HF, it may occur in patients with abdominal obesity or ascites and patients with pulmonary disease whose lung mechanics favor an upright posture.
 Paroxysmal nocturnal dyspnea (PND)
Paroxysmal nocturnal dyspnea (PND)
This term refers to acute episodes of severe shortness of breath and coughing that generally occur at night and awaken the patient from sleep, usually 1–3 h after the patient retires. PND may be manifest by coughing or wheezing, possibly because of increased pressure in the bronchial arteries leading to airway compression, along with interstitial pulmonary edema that leads to increased airway resistance. Whereas orthopnea may be relieved by sitting upright at the side of the bed with the legs in a dependent position, patients with PND often have persistent coughing and wheezing even after they have assumed the upright position. Cardiac asthma is closely related to PND, is characterized by wheezing secondary to bronchospasm, and must be differentiated from primary asthma and pulmonary causes of wheezing.
 Cheyne-Stokes respiration
Cheyne-Stokes respiration
Also referred to as periodic respiration or cyclic respiration, Cheyne-Stokes respiration is present in 40% of patients with advanced HF and usually is associated with low cardiac output. Cheyne-Stokes respiration is caused by a diminished sensitivity of the respiratory center to arterial Pco2. There is an apneic phase, during which arterial Po2 falls and arterial Pco2 rises. These changes in the arterial blood gas content stimulate the depressed respiratory center, resulting in hyperventilation and hypocapnia, followed by recurrence of apnea. Cheyne-Stokes respirations may be perceived by the patient or the patient’s family as severe dyspnea or as a transient cessation of breathing.
 Acute pulmonary edema
Acute pulmonary edema
See Chap. 28.
Other symptoms
Patients with HF also may present with gastrointestinal symptoms. Anorexia, nausea, and early satiety associated with abdominal pain and fullness are common complaints and may be related to edema of the bowel wall and/or a congested liver. Congestion of the liver and stretching of its capsule may lead to right-upperquadrant pain. Cerebral symptoms such as confusion, disorientation, and sleep and mood disturbances may be observed in patients with severe HF, particularly elderly patients with cerebral arteriosclerosis and reduced cerebral perfusion. Nocturia is common in HF and may contribute to insomnia.
PHYSICAL EXAMINATION
A careful physical examination is always warranted in the evaluation of patients with HF. The purpose of the examination is to help determine the cause of HF as well as to assess the severity of the syndrome. Obtaining additional information about the hemodynamic profile and the response to therapy and determining the prognosis are important additional goals of the physical examination.
General appearance and vital signs
In mild or moderately severe HF, the patient appears to be in no distress at rest except for feeling uncomfortable when lying flat for more than a few minutes. In more severe HF, the patient must sit upright, may have labored breathing, and may not be able to finish a sentence because of shortness of breath. Systolic blood pressure may be normal or high in early HF, but it generally is reduced in advanced HF because of severe LV dysfunction. The pulse pressure may be diminished, reflecting a reduction in stroke volume. Sinus tachycardia is a nonspecific sign caused by increased adrenergic activity. Peripheral vasoconstriction leading to cool peripheral extremities and cyanosis of the lips and nail beds is also caused by excessive adrenergic activity.
Jugular veins
(See also Chap. 9) Examination of the jugular veins provides an estimation of right atrial pressure. The jugular venous pressure is best appreciated with the patient lying recumbent, with the head tilted at 45°. The jugular venous pressure should be quantified in centimeters of water (normal ≤8 cm) by estimating the height of the venous column of blood above the sternal angle in centimeters and then adding 5 cm. In the early stages of HF, the venous pressure may be normal at rest but may become abnormally elevated with sustained (~1 min) pressure on the abdomen (positive abdominojugular reflux). Giant v waves indicate the presence of tricuspid regurgitation.
Pulmonary examination
Pulmonary crackles (rales or crepitations) result from the transudation of fluid from the intravascular space into the alveoli. In patients with pulmonary edema, rales may be heard widely over both lung fields and may be accompanied by expiratory wheezing (cardiac asthma). When present in patients without concomitant lung disease, rales are specific for HF. Importantly, rales are frequently absent in patients with chronic HF, even when LV filling pressures are elevated, because of increased lymphatic drainage of alveolar fluid. Pleural effusions result from the elevation of pleural capillary pressure and the resulting transudation of fluid into the pleural cavities. Since the pleural veins drain into both the systemic and the pulmonary veins, pleural effusions occur most commonly with biventricular failure. Although pleural effusions are often bilateral in HF, when they are unilateral, they occur more frequently in the right pleural space.
Cardiac examination
Examination of the heart, although essential, frequently does not provide useful information about the severity of HF. If cardiomegaly is present, the point of maximal impulse (PMI) usually is displaced below the fifth intercostal space and/or lateral to the midclavicular line, and the impulse is palpable over two interspaces. Severe LV hypertrophy leads to a sustained PMI. In some patients, a third heart sound (S3) is audible and palpable at the apex. Patients with enlarged or hypertrophied right ventricles may have a sustained and prolonged left parasternal impulse extending throughout systole. An S3 (or protodiastolic gallop) is most commonly present in patients with volume overload who have tachycardia and tachypnea, and it often signifies severe hemodynamic compromise. A fourth heart sound (S4) is not a specific indicator of HF but is usually present in patients with diastolic dysfunction. The murmurs of mitral and tricuspid regurgitation are frequently present in patients with advanced HF.
Abdomen and extremities
Hepatomegaly is an important sign in patients with HF. When it is present, the enlarged liver is frequently tender and may pulsate during systole if tricuspid regurgitation is present. Ascites, a late sign, occurs as a consequence of increased pressure in the hepatic veins and the veins draining the peritoneum. Jaundice, also a late finding in HF, results from impairment of hepatic function secondary to hepatic congestion and hepatocellular hypoxemia and is associated with elevations of both direct and indirect bilirubin.
Peripheral edema is a cardinal manifestation of HF, but it is nonspecific and usually is absent in patients who have been treated adequately with diuretics. Peripheral edema is usually symmetric and dependent in HF and occurs predominantly in the ankles and the pretibial region in ambulatory patients. In bedridden patients, edema may be found in the sacral area (presacral edema) and the scrotum. Long-standing edema may be associated with indurated and pigmented skin.
Cardiac cachexia
With severe chronic HF, there may be marked weight loss and cachexia. Although the mechanism of cachexia is not entirely understood, it is probably multifactorial and includes elevation of the resting metabolic rate; anorexia, nausea, and vomiting due to congestive hepatomegaly and abdominal fullness; elevation of circulating concentrations of cytokines such as TNF; and impairment of intestinal absorption due to congestion of the intestinal veins. When present, cachexia augurs a poor overall prognosis.
DIAGNOSIS
The diagnosis of HF is relatively straightforward when the patient presents with classic signs and symptoms of HF; however, the signs and symptoms of HF are neither specific nor sensitive. Accordingly, the key to making the diagnosis is to have a high index of suspicion, particularly for high-risk patients. When these patients present with signs or symptoms of HF, additional laboratory testing should be performed.
Routine laboratory testing
Patients with new-onset HF and those with chronic HF and acute decompensation should have a complete blood count, a panel of electrolytes, blood urea nitrogen, serum creatinine, hepatic enzymes, and a urinalysis. Selected patients should have assessment for diabetes mellitus (fasting serum glucose or oral glucose tolerance test), dyslipidemia (fasting lipid panel), and thyroid abnormalities (thyroid-stimulating hormone level).
Electrocardiogram (ECG)
A routine 12-lead ECG is recommended. The major importance of the ECG is to assess cardiac rhythm and determine the presence of LV hypertrophy or a prior MI (presence or absence of Q waves) as well as to determine QRS width to ascertain whether the patient may benefit from resynchronization therapy (see later). A normal ECG virtually excludes LV systolic dysfunction.
Chest x-ray
A chest x-ray provides useful information about cardiac size and shape, as well as the state of the pulmonary vasculature, and may identify noncardiac causes of the patient’s symptoms. Although patients with acute HF have evidence of pulmonary hypertension, interstitial edema, and/or pulmonary edema, the majority of patients with chronic HF do not. The absence of these findings in patients with chronic HF reflects the increased capacity of the lymphatics to remove interstitial and/or pulmonary fluid.
Assessment of LV function
Noninvasive cardiac imaging (Chap. 12) is essential for the diagnosis, evaluation, and management of HF. The most useful test is the two-dimensional (2-D) echocardiogram/Doppler, which can provide a semi-quantitative assessment of LV size and function as well as the presence or absence of valvular and/or regional wall motion abnormalities (indicative of a prior MI). The presence of left atrial dilation and LV hypertrophy, together with abnormalities of LV diastolic filling provided by pulse-wave and tissue Doppler, is useful for the assessment of HF with a preserved EF. The 2-D echocardiogram/Doppler is also invaluable in assessing RV size and pulmonary pressures, which are critical in the evaluation and management of cor pulmonale (see later). Magnetic resonance imaging (MRI) also provides a comprehensive analysis of cardiac anatomy and function and is now the gold standard for assessing LV mass and volumes. MRI also is emerging as a useful and accurate imaging modality for evaluating patients with HF, both in terms of assessing LV structure and for determining the cause of HF (e.g., amyloidosis, ischemic cardiomyopathy, hemochromatosis).
The most useful index of LV function is the EF (stroke volume divided by end-diastolic volume). Because the EF is easy to measure by noninvasive testing and easy to conceptualize, it has gained wide acceptance among clinicians. Unfortunately, the EF has a number of limitations as a true measure of contractility, since it is influenced by alterations in afterload and/or preload. Nonetheless, with the exceptions indicated above, when the EF is normal (≥50%), systolic function is usually adequate, and when the EF is significantly depressed (<30–40%), contractility is usually depressed.
Biomarkers
Circulating levels of natriuretic peptides are useful adjunctive tools in the diagnosis of patients with HF. Both B-type natriuretic peptide (BNP) and N-terminal pro-BNP, which are released from the failing heart, are relatively sensitive markers for the presence of HF with depressed EF; they also are elevated in HF patients with a preserved EF, albeit to a lesser degree. However, it is important to recognize that natriuretic peptide levels increase with age and renal impairment, are more elevated in women, and can be elevated in right HF from any cause. Levels can be falsely low in obese patients and may normalize in some patients after appropriate treatment. At present, serial measurements of BNP are not recommended as a guide to HF therapy. Other biomarkers, such as troponin T and I, C-reactive protein, TNF receptors, and uric acid, may be elevated in HF and provide important prognostic information. Serial measurements of one or more biomarkers ultimately may help guide therapy in HF, but they are not currently recommended for this purpose.
Exercise testing
Treadmill or bicycle exercise testing is not routinely advocated for patients with HF, but either is useful for assessing the need for cardiac transplantation in patients with advanced HF (Chap. 18). A peak oxygen uptake (Vo2) <14 mL/kg per min is associated with a relatively poor prognosis. Patients with a Vo2 <14 mL/kg per min have been shown, in general, to have better survival when transplanted than when treated medically.
DIFFERENTIAL DIAGNOSIS
HF resembles but should be distinguished from (1) conditions in which there is circulatory congestion secondary to abnormal salt and water retention but in which there is no disturbance of cardiac structure or function (e.g., renal failure) and (2) noncardiac causes of pulmonary edema (e.g., acute respiratory distress syndrome). In most patients who present with classic signs and symptoms of HF, the diagnosis is relatively straightforward. However, even experienced clinicians have difficulty differentiating the dyspnea that arises from cardiac and pulmonary causes (Chap. 5). In this regard, noninvasive cardiac imaging, biomarkers, pulmonary function testing, and chest x-ray may be useful. A very low BNP or N-terminal pro-BNP may be helpful in excluding a cardiac cause of dyspnea in this setting. Ankle edema may arise secondary to varicose veins, obesity, renal disease, or gravitational effects. When HF develops in patients with a preserved EF, it may be difficult to determine the relative contribution of HF to the dyspnea that occurs in chronic lung disease and/or obesity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


