1990 American College of Rheumatology classification criteria for Wegener’s granulomatosis
For purposes of classification, a patient shall be said to have Wegener’s granulomatosis if at least 2 of these 4 criteria are present. The presence of any 2 or more criteria yields a sensitivity of 88.2 % and a specificity of 92.0 %
1. Nasal or oral inflammation: Development of painful or painless oral ulcers or purulent or bloody nasal discharge
2. Abnormal chest radiograph: Chest radiograph showing the presence of nodules, fixed infiltrates, or cavities
3. Urinary sediment: Microhematuria (>5 red blood cells per high power field) or red cell casts in urine sediment
4. Granulomatous inflammation on biopsy: Histologic changes showing granulomatous inflammation within the wall of an artery or in the perivascular or extravascular area (artery or arteriole)
Definition of Wegener’s granulomatosis in the nomenclature of systemic vasculitis adopted in 1994 by the Chapel Hill consensus conference
Large-vessel vasculitis: Giant-cell (temporal) arteritis; Takayasu’s arteritis
Medium-sized-vessel vasculitis: Polyarteritis nodosa; Kawasaki’s disease
Small–vessel vasculitis:
Wegener’s granulomatosis*
Granulomatous inflammation involving the respiratory tract and necrotizing vasculitis affecting small to medium–sized vessels, e.g., capillaries, venules, arterioles, and arteries
Necrotizing glomerulonephritis is common
Churg-Strauss syndrome*
Microscopic polyangiitis*
Henoch-Schönlein purpura
Cryoglobulinemic vasculitis
Cutaneous leukocytoclastic angiitis
Small artery refers to distal arterial radicals that connect with arterioles. Small vessels include small arteries, arterioles, venules and capillaries
* These vasculitides are associated with ANCA
Definition of granulomatosis with polyangiitis (Wegener’s) in the 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides
Large vessel vasculitis: Giant-cell arteritis; Takayasu arteritis
Medium vessel vasculitis: Polyarteritis nodosa; Kawasaki disease
Small vessel vasculitis:
ANCA associated vasculitis
Microscopic polyangiitis
Granulomatosis with polyangiitis (Wegener’s)
Necrotizing granulomatous inflammation usually involving the upper and lower respiratory tract, and necrotizing vasculitis affecting predominantly small to medium vessels (e.g., capillaries, venules, arterioles, arteries and veins). Necrotizing glomerulonephritis is common
Eosinophilic granulomatosis with polyangiitis (Churg Strauss)
Immune complex vasculitis
Anti-GBM disease cryoglobulinemic
Vasculitis IgA vasculitis (Henoch-Schönlein)
Hypocomplementemic urticarial vasculitis (anti-C1q vasculitis)
Variable vessel vasculitis: Cogan’s syndrome; Behçet’s disease
Single organ vasculitis
Vasculitis associated with systemic disease
Vasculitis associated with probable etiology
Large vessels are the aorta and its major branches and the analogous veins. Medium vessels are the main visceral arteries and veins and their initial branches. Small vessels are intra–parenchymal arteries, arterioles, capillaries, venules and veins
Epidemiology
GPA affects both genders equally. Only a few studies, mostly those of localized/limited GPA, have suggested a relatively greater frequency in women. The median age at diagnosis is in the fifth decade (Table 9.2), but young children and older adults can be affected. Most patients (93–98 %) are white (Caucasian and Hispanics). The estimated annual incidence is 2–12 cases per million population and the prevalence 23–160 cases per million population [3]. A north-south gradient is suggested, at least in Europe, because the reported annual incidence is twice as high in Norway, for example, than in Spain (10.6 vs. 4.9 per million inhabitants) [11]. Conversely, antiPR3+ GPA is rare in Japan, where anti-myeloperoxidase (antiMPO) + disease (mostly microscopic polyangiitis-type) represents most cases of ANCA vasculitis. Notably, the incidence of the disease seems also to have increased within the past decades, according to several European studies, although whether these changes could be related, at least in part, to a better understanding of the disease and thus lead to more frequent and earlier diagnosis, especially since the discovery of ANCA in 1985, remains controversial [12]. A recent British study suggested peaks of incidence every 8–10 years (17.4 per year per million population during peaks vs. only 4.53 per year per million population not during peaks) [11]. Seasonal variations in GPA incidence have been reported, but with conflicting results.
Table 9.2
Characteristics of patients with granulomatosis with polyangiitis (Wegener) and frequency (percent), according to the main studies published between 1958 and 2011
Characteristic | Range | Mean |
|---|---|---|
Mean age at diagnosis (years) | 14–58 | 48 |
Clinical presentations/organ involvement (%) | ||
Ear, nose and throat | 56–99 | 70 |
Kidney | 18–100 | 58 |
Lung | 40–100 | 57 |
Arthralgias | 15–77 | 52 |
Fever | 17–72 | 45 |
Eye | 2–61 | 34 |
Skin | 12–50 | 29 |
Peripheral nervous system | 7–68 | 20 |
Heart | 0–30 | 13 |
Gastrointestinal | 0–42 | 12 |
Central nervous system | 0–13 | 8 |
The existence of these potential geographic and temporal variations in GPA incidence suggest a potential pathogenic, or at least participating, role of environment (allergic, chemical and/or infectious) and/or genetic factors in the development of the disease. The association of GPA and silica exposure, industrial pollutants such as cadmium, mercury derivatives or other heavy metals such as lead, volatile hydrocarbons or organic solvents has been reported. Other studies have suggested links between GPA and the inhalation of dust, especially during livestock activities [13]. However, GPA does not seem more frequent in rural areas, and exposure to such environmental agents is found in no more than 10 % of all GPA patients [14]. Finally, an inverse relation between the intensity of sun exposure, specifically ultraviolet rays, and GPA prevalence suggests a possible link with vitamin D deficiency, as has been suggested in many other autoimmune diseases [15].
GPA is not an inherited or genetic disease. Familial forms are extremely rare, with a small and insignificant relative risk of GPA among first-degree relatives of GPA patients (hazard ratio [HR] 1.56, 95 % confidence interval [95 % CI] 0.35–6.90), as compared with the general population. However, first-degree relatives may be more likely to develop other autoimmune diseases (HR 1.32, 1.18–1.49), including multiple sclerosis (HR 1.92), Sjögren’s syndrome (HR 2.00) or rheumatoid arthritis (HR 1.54) [16]. Personal (and probably also familial) history of autoimmune thyroiditis has been found more frequently in GPA than in the general population (13 % of GPA patients) [17]. Factors related to genetic predisposition are thus likely, although not enough to explain or trigger the disease by themselves. Several international teams are conducting studies on genome-wide associations with GPA.Many variable genetic associations have indeed been reported, the two most reproducible being those for molecules of major histocompatibility complex (MHC) HLA–DPB1*0401 (odds ratio [OR] 3.38 for patients with ANCA) and, to a lesser degree, allele deficiency of alpha-1 antitrypsin (serpin A1; PI*Z alleles in 5–27 % of GPA patients, PI*S alleles in 11.58 %, homozygosity for deficiency ZZ, SS or SZ having more severe forms) [18]. Many other genetic associations have been reported, including certain alleles of PR3-coding genes (−564 A/G), type IIa and IIIa/b Fc-gamma or -alpha receptors, intracellular tyrosine phosphatase PTPN22 (620 W allele), transforming growth factor-beta 1, interleukin-10 (IL-10) promoter, CTLA-4, or CD226 (Gly307Ser) polymorphism allele [18, 19]. GPA has been associated with other MHC molecules, including DR2 and DR4 allele HLA-DRB1*04, B8, DR1-DQw1, B50-DR9 and DR9 in Japanese patients. Conversely, the DR13-DR6 phenotype was found to be less frequent among Norwegians with GPA than healthy subjects.
More importantly perhaps, results of recent studies showed that genetic susceptibility was more linked with the ANCA type (antiPR3 with HLA–DP and the genes encoding α1-antitrypsin (SERPINA1) and proteinase 3 (PRTN3); antiMPO with HLA–DQ) than with the clinical phenotype (GPA versus microscopic polyangiitis) [20]. Whether the classification of ANCA-associated vasculitides should be modified according to these results is now under debate.
Pathogenesis
GPA is considered an autoimmune inflammatory disease. Defining its pathogenic mechanisms has advanced enormously within the past three decades, especially since the discovery of ANCA in 1985 [12]. However, the primum movens of the disease remain(s) to be identified [5, 6].
The hypothesis of an infectious agent, such as Staphy-lococcus aureus, (over)activating the immune system has been repeatedly suggested. Chronic nasal carriage of S. aureus is considered a risk factor for relapse, as is observed in some but not all patients and shown in one study, possibly by maintaining a local inflammatory immune response within the nasal mucosa [21]. A selective cross-reactivity of T cells towards PR3 and S. aureus antigens has been suggested. The experimental model of Pendergraft et al. suggested that some antigenic motives of S. aureus have a molecular similarity with the protein synthesized from the complementary DNA segment coding for human PR3, which can trigger the production of antibodies against PR3 by a protein-complementary idiotype–anti-idiotype mechanism [22]. The S. aureus infection found in GPA patients is not from a particular strain and does not produce specific toxins or lead to a specific T-cell repertoire selection through superantigenic mechanisms [23]. Other organisms could also be involved. Recently, anti-lysosomal membrane protein 2 (antiLAMP2), another and newer type of ANCA, was detected by Kain and colleagues in more than 90 % of patients with ANCA-related (antiPR3 as well as antiMPO) glomerulonephritis [24]. This group further showed the LAMP-2 epitope with 100 % homology to the bacterial adhesin FimH located at the tip of type 1 fimbriae and crucial for attachment to host epithelia of Gram-negative pathogens such as Escherichia coli, Klebsiella pneumoniae and Proteus mirabilis. In addition, pauci-immune glomerulonephritis developed in rats immunized with FimH. However, those results have not been replicated and thus remain controvertial [25].
Whatever the signal for their synthesis, PR3-ANCA are detected in more than 80 % of patients with generalized GPA. However, their pathogenic role is less well documented than that of pANCA antiMPO (most characteristic of microscopic polyangiitis). Until recently, convincing animal models of vasculitis associated with antiPR3 ANCA were lacking as compared with those associated with antiMPO. In the mouse model of Pfister et al., vasculitis induced by transfer of antiPR3 ANCA remained localized to the mouse footpads, was not granulomatous and required prior sensitization with subcutaneous injections of tumor necrosis factor a (TNF-α) [26]. In the BALB/c murine model of Pendergraft et al., mice did not develop overt vasculitis [22]. More recently, two murine models of antiPR3 ANCA-associated vasculitis have been developed and are more convincing but require specific genetic backgrounds and prior subtle and complicated immune manipulations (including the humanization of the mouse immune system because of the lack of PR3 expression in murine neutrophils and low human and murine PR3 homology) [27, 28]. Specific alterations and “maturation” may be necessary for antiPR3 ANCA to become pathogenic, including the selection of higher-affinity ANCA in the nasal mucosa granulomas or modulation of their sialylation levels [29]. Although the pathogenicity of anti-PR3 ANCA remains difficult to demonstrate, results from recent studies of therapies targeting B cells (i.e., rituximab) provide indirect evidence for their potential role, unless these biologic agents act through other more complex pathways [30–32].
Other factors or mechanisms involved in GPA can favor or enhance the PR3-antiPR3 ANCA immune response or act independently. Besides the frequent functional and/or genetic deficit in α-1-antitrypsin, the physiological inhibitor of PR3, overexpression of PR3 on the neutrophil membrane, genetically determined, has also been reported. More recently, circulating microparticles derived from platelets, neutrophils or endothelial cells, as well as neutrophil extracellular traps (cellular activation debris produced in response to inflammatory signals), were found to express PR3 and MPO, thus perpetuating the presentation of self-antigens, and could trigger or increase inflammatory responses in endothelia, especially in kidneys [33, 34]. GPA has been associated with excess production of certain soluble factors of stimulation and proliferation of B lymphocytes (B-cell activating factor of the TNF family, also called B-lymphocyte stimulator). In parallel, functional abnormalities of the regulatory T lymphocytes CD4+ CD25+ FoxP3+ and abnormal expression of certain T-cell co-stimulatory molecules, including increased membrane expression of CTLA4, can lead to rupture of immune tolerance mechanisms.
The binding of ANCA results in activation of the alternative complement pathway and production of reactive oxygen species by neutrophils, all of which lead to endothelial injury (depletion of neutrophils or blockade of complement pathway both prevent the development of antiMPO-induced vasculitis in experimental models) [35, 36]. Granuloma formation, an important histologic characteristic of GPA, involves lymphocyte subpopulations, with a preferential cytokine secretion profile of TH1 lymphocytes (interferon-gamma), but also other complex cytokine imbalances, cell populations or immune pathways of more recent discovery, such as TH17 lymphocytes, a source of IL-17, dendritic or natural killer cells. Functional abnormalities of endothelial cells have been described, and serum autoantibodies against endothelial cells have been found in some patients.
Clinical Manifestations
The main clinical manifestations of the GPA and their frequencies are in Table 9.2. The most frequent target organs or systems are the upper and lower respiratory tracts and kidneys (necrotizing crescentic pauci-immune glomerulonephritis), but any organ can be affected. Complications of treatment and disease damage are discussed in another section of this chapter.
Constitutional Symptoms
Constitutional and musculoskeletal symptoms are common and include asthenia, fever, weight loss, diffuse myalgias, arthralgias, or sometimes genuine inflammatory arthritis with reported cases of oligo- or polyarthritis.
Ear, Nose and Throat Manifestations
More than two-thirds of the patients have ear, nose and throat manifestations, which often represent the first symptoms of the disease. When isolated, such symptoms can result in diagnostic delay. Persistent nasal obstruction, nasal or sinus pain, sinusitis, rhinitis, recurrent epistaxes and/or nasal crusting, or serous otitis media and/or hypoacousia should alert physicians to the possibility of GPA [4]. Hyposmia and/or hypogeusia are frequent.
The destruction of the nasal cartilage, which can lead to nasal septum perforation and/or saddle-nose bridge deformity (Fig. 9.1), is very suggestive although not pathognomonic of GPA [37, 38]. The cartilage of the ears can also be affected (chondritis), as can osteochondral tissues of the face and skull, with rare occurrence of palate perforation or development of fistulas between sinus and orbital cavities.
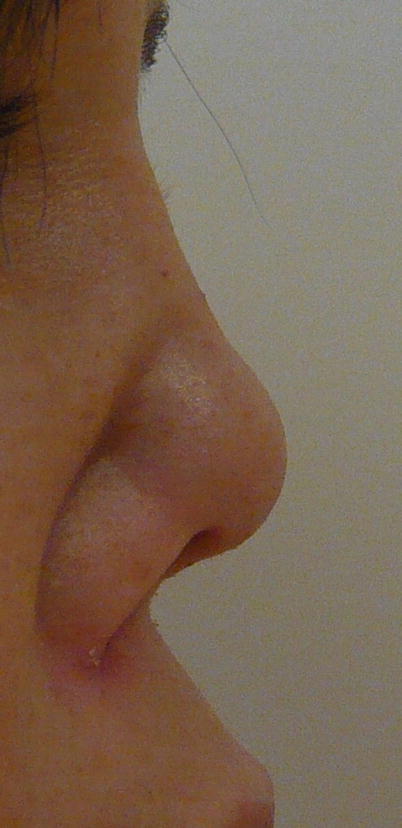
Fig. 9.1
Nasal deformity (bridge erosion) in a patient with granulomatosis with polyangiitis (Wegener)
Another classic but rarer upper-respiratory-tract lesion (7–15 % of patients) is subglottic stenosis, which is responsible for dysphonia, dyspnea, with or without stridor, and may require emergency procedures (dilatation with local injections of corticosteroids or tracheostomy) [38, 39]. Subglottic stenosis can be associated with endobronchial stenoses or can be isolated. It can parallel other manifestations or continuously progress despite control of disease elsewhere [40].
CT scan of the sinuses may show unilateral or bilateral sinusitis, osteochondral destruction and/or osteosclerosis (Fig. 9.2), otitis media and/or mastoiditis. Granulomatous inflammatory pseudotumors can also occur and can infiltrate the sinuses, skull base and/or orbits and be responsible for pain, proptosis, or contiguous pachymeningitis, which incur risk of compression of surrounding structures, such as cranial (ophthalmoplegia) or optic nerves. CT scan findings of Subglottic stenosis should be studied in parallel with results of a careful endoscopy (biopsies of subglottic stenosis are relatively sensitive but risky). Biopsies of nasal and/or sinus lesions can reveal granulomatous inflammation or vasculitis in about half of cases, when sufficiently deep in and under the mucosa [41, 42]. In routine practice, nasal and sinus biopsies are often superficial and rarely contribute to diagnosis (abnormal in <25 % of cases).
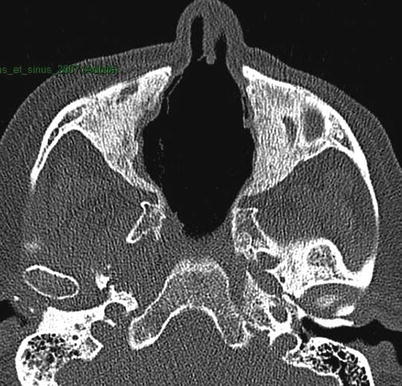
Fig. 9.2
Sinus CT scan (horizontal) in a patient with granulomatosis with polyangiitis (Wegener). Major destruction of septum and midline cartilaginous structures, along with bilateral maxillary sinus osteosclerosis and atrophy
Pulmonary Manifestations
Lungs are involved in 70–100 % of patients, with clinical manifestations ranging from mild cough, dyspnea, chest pain, and intermittent hemoptoic expectoration to acute respiratory distress syndrome due to massive alveolar hemorrhage. In 6 % of cases, lung involvement can remain asymptomatic, especially with lung nodules.
Lung nodules are among the most characteristic signs. Chest X-ray and CT scan can show nodules in 40–66 % of patients [43]; nodules are unilateral or bilateral, single or multiple (generally <10), measuring 0.5–10 cm in diameter, and excavated in half of cases (Figs. 9.3, 9.4, 9.5, and 9.6). High-resolution chest CT should be obtained for all patients with respiratory symptoms because small nodules, early alveolar hemorrhage and other early lesions may be missed in chest X-ray. The differential diagnosis, including primary or metastatic tumors and tuberculosis or fungal infections, can be difficult.
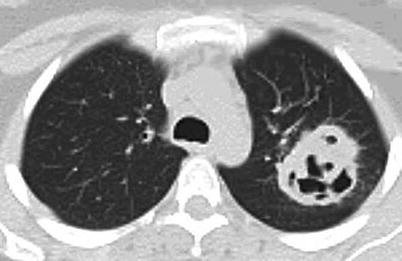
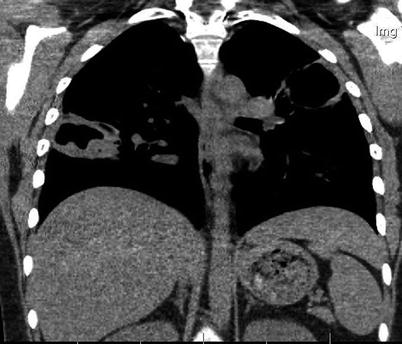
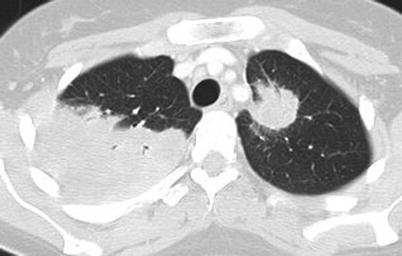
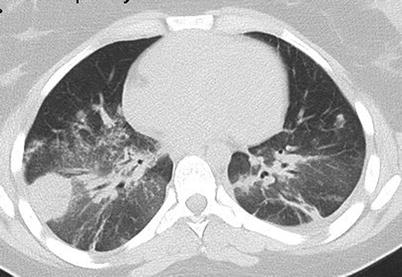
Fig. 9.3
Chest CT scan (horizontal) in a patient with granulomatosis with polyangiitis (Wegener). Large parenchymal excavated nodule (left lung)
Fig. 9.4
Chest CT scan (coronal) in a patient with granulomatosis with polyangiitis (Wegener). Large parenchymal excavated nodules in both lungs
Fig. 9.5
Chest CT scan (horizontal) in a patient with granulomatosis with polyangiitis (Wegener). Parenchymal plain nodule (left lung) associated with right lung posterior consolidation (and mild pleural effusion)
Fig. 9.6
Chest CT scan (horizontal) in a patient with granulomatosis with polyangiitis (Wegener). Parenchymal plain nodule (right lung) associated with multiple bilateral patchy opacities and diffuse posterior ground glass opacities (probably corresponding to moderate alveolar hemorrhage)
Alveolar hemorrhage (8–30 % of patients) can occur at symptom onset or later and be associated with lung nodules [4]. It can be limited to a few bloody expectorations or become rapidly massive and be responsible for acute respiratory failure. However, sometimes hemorrhage is suspected only on chest CT scan (Figs. 9.6, 9.7, and 9.8; patchy, ground-glass opacities) and/or as unexplained anemia, then confirmed by broncho-alveolar lavage (demonstrating persistently hemorrhagic fluid on sequential samples). Even when overt, broncho-alveolar lavage should be considered to rule out concurrent infection, even at disease onset.
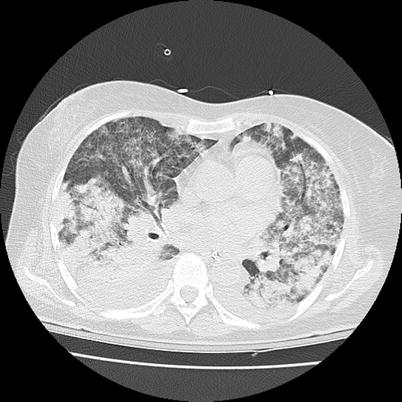
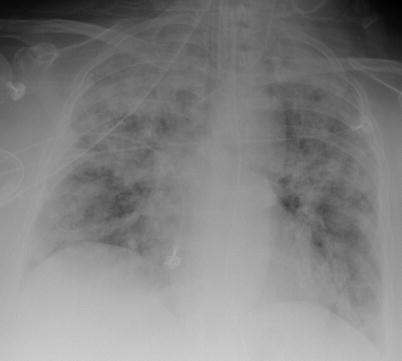
Fig. 9.7
Chest CT scan (horizontal) in a patient with granulomatosis with polyangiitis (Wegener). Diffuse bilateral ground glass infiltrate corresponding to massive alveolar hemorrhage
Fig. 9.8
Chest radiograph (anteroposterior) in a patient with granulomatosis with polyangiitis (Wegener). Diffuse bilateral bronchoalveolar infiltrate corresponding to massive alveolar hemorrhage
Other pulmonary infiltrates or lung consolidation (Fig. 9.5), unilateral or bilateral, may be observed in 30–50 % of patients and pleural effusion in 9–28 % [4, 43]. Spontaneous pneumothorax or pyopneumothorax is rare but can occur. Bronchial stenoses, usually on main bronchia and/or first branches, are uncommon and are often difficult to manage, as mentioned previously (frequently associated with subglottic stenosis). These are best studied with CT scan of lungs (Fig. 9.9) and fiberoptic bronchoscopy, which can reveal multifocal strictures and/or granulomatous endobronchial lesions.
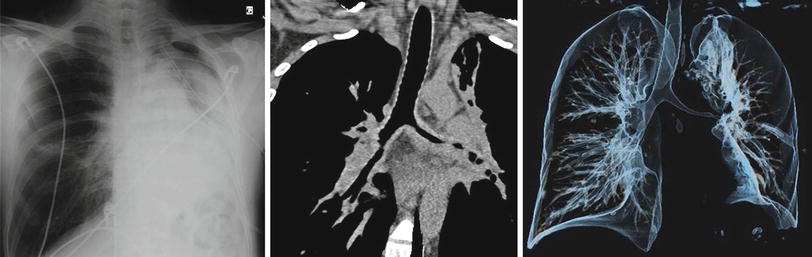
Fig. 9.9
Chest radiograph (anteroposterior), CT scan (coronal) and 3D-volume reconstruction of the tracheobronchial tree in a patient with granulomatosis with polyangiitis (Wegener). Major lengthy and multisegmental stenosis of the left main bronchus causing partial left lung atelectasis (bronchus intermedius is also narrowed laterally). Note the thickening of the narrowed bronchial wall, suggesting granulomatous infiltration
Surgical and open-wedged lung biopsies, targeting nodules or consolidation lesions, have good diagnostic yield, up to 91 % [4, 42]. In practice, obtaining these biopsies can be difficult, but they can be necessary to confirm the diagnosis, especially in patients with disease limited to the lungs.
Pulmonary embolism (<5 % of patients at diagnosis) should be considered a sign of active GPA, especially in patients with otherwise unexplained chest pain and/or sudden shortness of breath.
Asthma is not a feature of GPA and may suggest eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome). Coincidental association of the two conditions remains possible, and an eosinophilic variant form of GPA has been described, sometimes with asthma. Chronic interstitial lung disease with fibrosis is not a classical feature of GPA, as compared to antiMPO-associated lung fibrosis with or without systemic vasculitis (microscopic polyangiitis), but has been reported.
Kidney and Urologic Manifestations
Kidney
Renal disease represents the third most frequent manifestation of GPA and presents as focal, segmental, crescentic, necrotizing and pauci-immune glomerulonephritis. The first sign is microscopic hematuria, with (or without) granular red blood cell casts. Increased proteinuria usually follows before renal function worsening, which can rapidly lead to end-stage renal disease. The combination of alveolar hemorrhage and renal disease defines pulmonary–renal syndrome, which can occur in GPA, as well as in microscopic polyangiitis, anti-glomerular basement membrane (GBM) antibody disease (Goodpasture’s syndrome) and systemic lupus erythematosus. Kidney biopsy is sensitive in patients with urine sediment abnormalities, and histology can help in predicting renal prognosis (with a simple classification: focal, crescentic, mixed, and sclerotic) [44].
Rarer renal manifestations include granulomatous nephritis, interstitial granulomatous renal masses and aneurysms of the branches of the renal arteries, including their intraparenchymal portions [45, 46]. A possible (but low) increase in frequency of kidney cancer has been suggested by some studies or case reports, but most were probably related to bladder-toxic treatments [47].
Urological Manifestations
Ureteral (or, less frequently, urethral) stenosis, uni- or bilateral, single or multifocal, secondary to ureteral arteriolitis, peri-ureteral granulomatous infiltrates and/or sheathing in retroperitoneal fibrosis can occur and cause hydronephrosis and/or obstructive renal insufficiency [46]. Granulomatous prostatis, ischemic and/or granulomatous orchitis, and penile ulcers have been reported [46]. Vulvar ulcerations have been reported in women.
Neurologic Manifestations
Peripheral Nervous System Manifestations
The peripheral nervous system is affected in 11–68 % of patients [50, 51]. Clinically, (asymmetrical and asynchronous) mononeuritis multiplex represents the principal pattern of peripheral nervous system involvement (45–79 % of cases), most frequently involving ulnar and peroneal nerves, followed by sensorimotor (symmetrical) polyneuropathy, both related to axonal ischemia due to vasculitis of the vasa nervorum of the small epineural vessels [50].
Central Nervous System Manifestations
The central nervous system is involved more rarely, in 6–13 % of patients, and often later and more progressively (except for the exceptional strokes) than the PNS [52]. Central nervous system involvement can result from extension of sinusal and/or orbital granulomatous lesions causing pachymeningitis and sometimes (post- or pan-) pituitary gland involvement (diabetes insipidus) or a new development of cerebral granulomatous lesions and/or intracranial artery vasculitis. Headache, meningeal irritation, cranial nerve palsy, hypoacousia, and sensorimotor deficit are the most frequent clinical features, but hemiparesis, hemiplegia or seizures can occur (usually ischemic). Rare cases of cerebral venous thrombosis with cortical venous infarction have been reported.
Spinal Cord and Cranial Nerve Involvement
Spinal cord or cauda equina involvement is rare, and usually due to compression by a meningeal granulomatous infiltrate rather than spinal cord ischemic vasculitis [53]. Cranial nerve involvements are more common (4–14 % of patients), primarily affecting the optic nerves (II), VI and/or VII and V, uni- or bilaterally and due to compression by extensive meningeal or pseudotumoral intraorbital lesions or, more rarely, nerve ischemia and/or inflammation.
Skin and Oral Mucosal Manifestations
In total, 10–50 % of patients show skin lesions, primarily palpable purpura (Fig. 9.10). Macules, papules, ulcers, digital gangrene or, more rarely, subcutaneous nodules can occur [54]. Involvement of the elbows and hands, including dorsal face and digital pulps, is common. Lesions can sometimes mimic erythema elevatum diutinum or pyoderma gangrenosum, which can also occur as a complication or an associated condition.
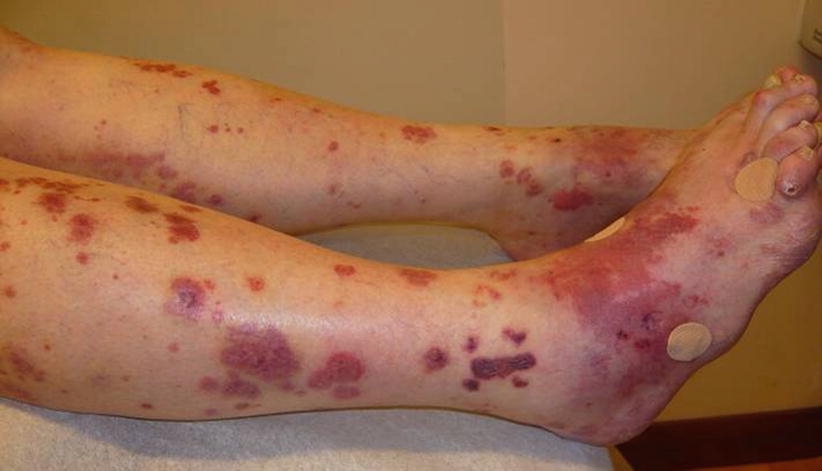
Fig. 9.10
Diffuse ecchymotic, necrotic and purpuric lesions in a patient with granulomatosis with polyangiitis (Wegener)
Skin biopsies most often reveal nonspecific perivascular infiltrates and/or aspects of leukocytoclastic vasculitis of small vessels, which is not specific to GPA. Sometimes blatant necrotizing vasculitis of superficial vessels of the dermis and/or deep subcutaneous layers is seen. Vascular or extravascular granulomatous infiltrates can be seen in nodular or papular lesions [55].
Oral mucosal lesions can occur in 10–50 % of patients and include ulcerations, persistent canker sores, especially on the lateral edges of the tongue, and gingival hypertrophy or “strawberry” gum, which is often painful and relatively evocative of GPA [54]. Infiltrations of parotid and/or accessory salivary glands have been described.
Eye Manifestations
Ocular and/or orbital manifestations are relatively common (14–60 % of patients) and can be inaugural and/or remain isolated for a long time.
Proptosis, possibly associated with ophthalmoplegia, is usually due to the local extension of a granulomatous retro-orbital pseudotumor or from ear, nose and throat and/or meningeal lesions (Fig. 9.11).
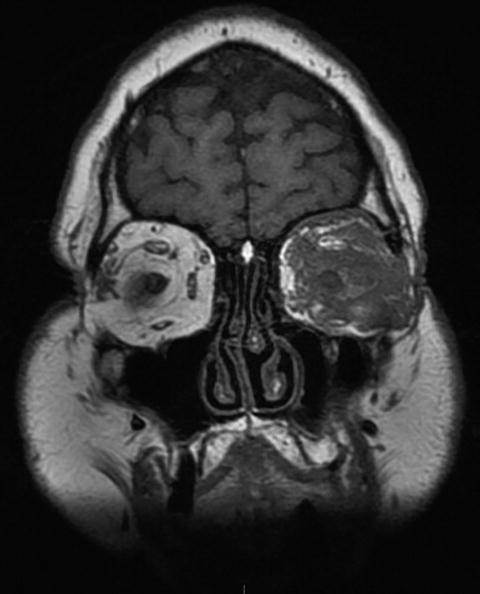
Fig. 9.11
Sinus and orbital CT scan (coronal) in a patient with granulomatosis with polyangiitis (Wegener). Massive infiltration of the left orbital cavity by a tumoral tissue (likely granulomatous on biopsy and exerting compression on eye and ocular muscles)
Conjunctivitis and episcleritis are relatively common and benign. Inflammation of the lacrimal gland (dacryocystitis) is also common and leads to a dry-eye sensation, watery eyes, or suprainfection because of clogged tear ducts, which may require surgical debridement procedure and/or stenting. Corneal ulcers and necrotizing nodular scleritis are more of a concern because of risk of eye perforation, loss of vision and endophthalmitis. Retinal vasculitis is rarer but can also cause blindness [56]. Extensive xanthelasma has been reported, especially after the regression of an orbital pseudotumor [54].
Cardiac Involvement
Cardiac involvement is rare (about 10 % of patients but up to 30 % in an autopsy series) [57]. MRI or sophisticated echocardiography (such as 2-D speckle-tracking) of the heart or electrophysiologic investigations can reveal subclinical abnormalities, whose prognostic value remain to be determined [58].
Sinus tachycardia is common during the active phases of the disease, as are arrhythmias, especially atrial fibrillation. Conduction disorders due to granulomatous infiltration of cardiac conductive tissue can lead to atrioventricular block or bundle branch block, which may require transient pacing but usually regresses with medical treatment. Pericarditis, sometimes progressing to tamponade and/or constrictive pericarditis, and coronary artery inflammation, most often silent clinically, account for half of the reported cardiac manifestations in GPA. Myocardial infarction, diagnosed during the patient’s life, represent about 10 % of the reported cases of GPA, with cardiac involvement and valvular disease, primarily aortic, in 21 % of such cases [59].
Gastrointestinal Manifestations
Gastrointestinal manifestations are less frequent in GPA than in other medium- and small-sized vessel vasculitides and are rarely isolated or present at disease onset; they range from mild abdominal pain to more severe ischemic bowel perforations [60]. Inflammatory granulomatous ileocolitis, gastritis or anorectitis are more characteristic but are not specific (differentiating between GPA, Crohn’s disease and ulcerative colitis can be difficult). Biopsies performed endoscopically rarely reveal vasculitis (10–50 %) and are not without risk.
The involvement of the appendix or pancreas (sometimes with challenging tumoral presentation) and/or gallbladder has been reported. Hepatic involvement is rare in clinical practice and usually remains limited to laboratory abnormalities (transaminitis). Splenic involvement is exceptional but can be responsible for infarction or non-traumatic rupture. Hepatic artery aneurysms, which can rupture, have been reported.
Gynecologic and Obstetric Manifestations
Few cases of mastitis have been described but can present as breast masses or ulcerated skin lesions. Uterine, adnexal and vulvar lesions are rare.
Few pregnancies in GPA patients have been reported in the literature, in part because the average age of onset of the disease is about 45 years and because cyclophosphamide, often used as treatment, can lead to infertility or subfertility in women of childbearing age, depending on the cumulative dose received [61]. Decreased fertility resulting from the disease itself is possible, but overt ovarian involvement has rarely been described. Measurement of anti-Mullerian hormone at treatment onset (and thereafter) can help evaluate the remaining follicles in young women.
Few women develop GPA during pregnancy and few others with known GPA experience disease flare or worsening during pregnancy post-partum or -abortion [62], some with fatal outcome. However, more than half of the reported pregnancies with GPA have been uneventful or with only minor disease manifestations. The risk of GPA worsening or relapse is an estimated 25 % if the disease is in remission at the onset of pregnancy and 40 % if the disease is active. The existence of organ damage from previous flares, especially renal and/or heart failure, must be taken into consideration when evaluating the risk with pregnancy. GPA flare and complications during pregnancy must be managed in referral centers for vasculitis and high-risk pregnancies.
Among pregnancies carried to term with GPA, newborn and child outcomes appear to be favourable, perhaps with a slightly higher frequency of preterm deliveries [62].
Venous Thrombosis and Other Vascular Events
A few studies have reproducibly demonstrated an increased risk of venous thromboembolic events (phlebitis and/or pulmonary embolism) with GPA, mainly during active phases of the disease [63, 64]. The incidence of these events was an estimated 7 per 100 patient-years in one of the first studies reporting this complication, which is a 20-fold higher risk than in the general population [64]. The events are probably favored by systemic and vessel wall inflammation and frequent reduced patient mobility because of the disease and/or neuropathy in some patients. Additional autoimmune mechanisms may lead to the development of these thromboses (antibodies against plasminogen have been detected in some patients, and cANCA may cross-react with plasminogen in certain conditions) [65].
Besides digital ischemia, stroke and/or coronary artery involvement, limb ischemia or carotid artery thromboses can occur, although rarely. A few cases of ascending aorta inflammation (aortitis), pseudo-tumor (periaortitis), or aneurysms of the aorta, subclavian arteries, popliteal, renal, hepatic and/or spleen have been reported. There is also a risk of ischemic heart disease, possibly because of endothelial function abnormalities, some of which are reversible in part with immunosuppressive therapy and prolonged use of corticosteroids [66].
Other Manifestations
Other rare manifestations include periosteitis, almost exclusively of the tibia, or pre-vertebral dorsal lesions, which can mimic fibrosing mediastinitis but are usually not erosive or compressive [67, 68]. Retroperitoneal fibrosis is exceptional.
Granulomatous involvement of the thyroid gland is rare, but auto-immune hypothyroidism (Hashimoto’s) or hyperthyroidism (Graves disease) can occur. Other endocrine glands that can be affected include adrenal and pituitary gland (as described with CNS manifestations). Pancreatic involvement does not usually result in secondary diabetes mellitus.
Limited/Localized Versus Severe/Diffuse/Systemic Forms
Despite variation in definitions among studies and authors (Table 9.3), GPA can be differentiated into two subgroups: generalized/diffuse/severe, characterized by the involvement of one or more major organ(s), including progressive renal disease and/or extensive alveolar hemorrhage, and limited/localized/early systemic, which predominantly presents as isolated ear, nose and throat diseases and is not directly life-threatening [17, 69–71]. Localized/limited forms account for up to 29 % of GPA at diagnosis, particularly in women, who are slightly younger than those with diffuse GPA (41 vs. 50 years old at diagnosis). ANCA are found in more than 90 % of patients with systemic/diffuse/severe GPA but only 50–78 % of those with limited forms. However, transition from a localized to a systemic form and vice versa is possible during the disease. Strictly and persistently localized GPA is exceptional (<5 % of patients in both German and French cohorts after 3 years of follow-up) [72, 73].
Table 9.3
Definitions of forms of granulomatosis with polyangiitis (Wegener)
Study group | Clinical subgroup | Systemic vasculitis outside ENT and lungs | Threatened vital organ function | Other definitions | Serum Creatinine (μmol/l) |
|---|---|---|---|---|---|
EUVAS (European group) | Localized | No | No | No constitutional symptoms, ANCA typically negative | <120 |
Early systemic | Yes | No | Constitutional symptoms present, ANCA-positive or -negative | <120 | |
Generalized | Yes | Yes | ANCA-positive | <500 | |
Severe | Yes | Organ failure | ANCA-positive | >500 | |
Refractory | Yes | Yes | Refractory to standard therapy | Any | |
WGET Research Group/VCRC (North American group) | Limited | Allowed, but not required | No | Not severe | ≤124, if hematuria, but no red blood cell casts |
Severe | Yes | Yes | Organ- or life-threatening disease, implies the need for cyclophosphamide (or rituximab) for remission-induction | Any |
Pediatric GPA
GPA is rare in children. Clinical manifestations are similar to that for adults, but girls are more frequently affected than are boys, and nasal deformities and subglottic stenosis are more frequent [74] (Table 9.4). Kidney damage is also frequent and often with poorer prognosis than in adults. Venous thrombosis may also occur more frequently (up to 16 % of children with GPA). In contrast, neurological manifestations seem less frequent (18 % of cases) [75]. However, the overall prognosis is similar but with significant morbidity because of damage from disease and treatment side effects.
Table 9.4
EULAR/PRINTO/PreS criteria and classification definition of granulomatosis with polyangiitis (Wegener) in children, Ankara 2008
Children GPA is a systemic inflammatory disease characterised by at least three of the six following criteria: | |
|---|---|
Criterion | Glossary |
1. Histopathology | Granulomatous inflammation within the wall of an artery or in the perivascular or extravascular area |
2. Upper airway involvement | Chronic purulent or bloody nasal discharge or recurrent epistaxis/crusts/granulomata, Nasal septum perforation or saddle nose deformity, Chronic or recurrent sinus inflammation |
3. Laryngo-tracheo-bronchial involvement | Subglottic, tracheal or bronchial stenoses |
4. Pulmonary involvement | Chest x-ray or CT showing the presence of nodules, cavities or fixed infiltrates |
5. ANCA | ANCA positivity by immunofluorescence or by ELISA (MPO/p or PR3/c ANCA) |
6. Renal involvement | Proteinuria >0.3 g/24 h or >30 mmol/mg of urine albumin/creatinine ratio on a spot morning sample, Hematuria or red blood cell casts: >5 red blood cells/high power field or red blood cells casts in the urinary sediment or ≥2+ on dipstick, Necrotising pauci-immune glomerulonephritis |
Diagnosis
Diagnostic Approach
Classification criteria have been defined (Table 9.1 for adults and Table 9.4 for children) but not diagnostic criteria. Efforts are ongoing internationally to revise existing classification criteria and, if possible, devise diagnostic ones. However, diagnosis of GPA is relatively simple in patients with typical clinical features (Box 9.1) such as a combination of nasal crusting and erosive rhinitis, lung nodules, renal involvement with microscopic hematuria and proteinuria, skin purpura on lower limbs and mononeuritis multiplex. The presence of antiPR3 cANCA will almost definitively support the diagnosis, without the need for biopsy. However, even in this setting, as well as in less obvious ones, histological confirmation may be necessary. Renal biopsy in the setting of renal impairment may also provide prognostic information for renal recovery [44]. In addition, several mimickers that must be ruled out include infections (mainly endocarditis) and cocaine use, which can be both (transiently) associated with antiPR3 ANCA.
Box 9.1
Main diagnostic features of granulomatosis with polyangiitis (GPA; Wegener’s granulomatosis) – there is presently no official and validated set of diagnostic criteria
Non–specific clinical and radiological features |
Constitutional symptoms: fever, arthralgias, myalgias, weight loss (in severe/generalized GPA) |
Skin purpuric lesions, sometimes necrotic (common to several medium and small vessel vasculitides) |
Mononeuritis multiplex (also common in several medium and small vessel vasculitides) |
More suggestive clinical and radiological features |
Signs of glomerulonephritis: microscopic hematuria (>5 red blood cells per high power field) with proteinuria and red cell casts in urine sediment (also in other small vessel vasculitides), then renal function impairment |
Ear nose and throat manifestations: nasal or oral ulcerations, recurrent serous otitis media, painful eroding sinusitis, purulent or bloody nasal discharge with nasal crusting, septum perforation and/or saddle nose deformity, subglottic stenosis |
Lung manifestations: alveolar hemorrhage (also in some other small vessel vasculitides), parenchymal nodules or cavities, bronchial stenosis |
Other “granulomatous-type” lesions: pachymeningitis, orbital (pseudo-)tumor |
Eye lesions: scleritis, dacrocystitis, retinal involvement |
Laboratory investigations to support the diagnosis |
Detection of serum ANCA (mainly, but not always and not exclusively, antiPR3 cANCA) in (50–75 % of the patients with limited GPA, and 80–90 % of those with severe/generalized GPA |
Evidence of granulomatous inflammation within the wall of a small artery or arteriole on biopsy of an affected organ/tissue with/without fibrinoid necrosis (diagnostic yield varies depending on which organ is biopsied – highest for open lung biopsies, lowest for nasal mucosa biopsies) |
Clinical Vignette
Four years earlier, a 50-year old female was hospitalized for severe anemia. She presented with dyspnea and initial laboratory tests revealed hemoglobin of 6.5 g/dL. She was having recurrent episodes of sinus and ear pain for the prior 6 months and intermittent nose bleeds. On exam, she had a nasal ulcer and bilateral chest rales. Her urine sediment analysis revealed microscopic hematuria and several red blood cell casts. Serum creatinine was 2.0 mg/dL (176 μmol/l), erythrocyte sedimentation rate was 78 mm/h. A chest computed tomography showed bilateral lung infiltrates and further tests revealed a positive anti-neutrophil cytoplasmic antibodies (ANCA) against proteinase 3 (PR3). A diagnosis of granulomatosis with polyangiitis (GPA – former Wegener’s granulomatosis) was made and treatment with cyclophosphamide and corticosteroid was initiated. Patient had excellent response to therapy and 4 months later, cyclophosphamide was switched to azathioprine. Her serum creatinine at that time was 1.0 mg/dL (88 μmol/l). Corticosteroid dose was slowly decreased over a year and discontinued. Patient remained asymptomatic for 2 years, and azathioprine was stopped.
She now presents for a routine follow-up visit. She is not taking any medications. She has polyarthralgias, bloody-tinged rhinorrhea, dry cough and bilateral ear fullness. Her serum hemoglobin is 12 g/dL and serum creatinine is 2.5 mg/dL (221 μmol/l). A diagnosis of GPA relapse is suspected.
Laboratory Investigations
Biology
Nonspecific inflammatory syndrome is common at diagnosis and during disease flare, with increased neutrophil count, normochromic normocytic anemia (50–73 % of patients, usually worse in patients with alveolar hemorrhage) and thrombocytosis (30–65 %) [4]. Eryrthocyte sedimentation rate in the first hour and C-reactive protein level are increased in almost every patient with active disease [76].
Transient and moderate eosinophilia can occur, in less than 12 % of patients and rarely above 2,000/mm3, sometimes with concomitant increase in serum IgE level. High levels of IgE and/or atypical findings, including asthma, should suggest eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome). Moderate lymphopenia, 700–750/mm3, is common at diagnosis (and during active disease) but can also result from and/or be induced by corticosteroid therapy [77].
Routine laboratory tests should systematically be part of the initial work-up and monitoring during the disease course. Analysis of fresh urine sediment is important for all patients for detecting erythrocyte casts, which are more suggestive of glomerular disease than a tubular or interstitial condition.
Immunology
The detection of cANCA (by indirect immunofluorescence) with antiPR3 (by ELISA) is a major tool in diagnosis. Other detection techniques have been developed, that may yield faster results and/or a better sensitivity [78]. However, the frequency is lower in patients with limited GPA (50–75 %) than generalized GPA (80–90 %). The prognostic value of ANCA and clinical utility of serial monitoring during treatment and thereafter are low and therefore should not dictate treatment adjustment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


