Chapter 5
Glycemia and CVD and Its Management
Diabetes, Glucose and Cardiovascular Disease
Cardiovascular disease (CVD) including coronary heart disease (CHD) is the major cause of mortality in patients with diabetes [1–3]. In patients without diabetes several well-studied environmental and physiological factors are now documented for CVD. These include hypercholesterolemia, hypertension, age, cigarette smoking, and obesity. Patients with diabetes are at a considerably higher risk [4] than those without and there are additive effects of other risk factors [5]. Of interest is that no more than 25% of the excess CHD risk in diabetes can be accounted for by established risk factors [6]. This illustrates the complexity of CVD, particularly in high-risk states such as diabetes, and may explain the inaccuracy of methods such as the Framingham equation and the PROCAM calculation to predict CHD risk in diabetes [7]. Until recently, the contribution of fluctuations and changes in glucose levels to CVD risk was relatively ignored. It should be noted that many of the CVD risk-prediction tools do not take into account glycemic control or fluctuations in glucose excursions. Our understanding of the direct and indirect effects of glucose on vascular dysfunction has grown over the past decade and basic scientific, epidemiological and clinical research has now demonstrated an independent association between glucose and CVD risk. For example, glucose-mediated oxidative stress is an important mediator of endothelial dysfunction [7]. Recent epidemiological data has confirmed associations between plasma glucose and CVD risk [3].
In recent years, there has been evidence to suggest that not only hyperglycemia contributes to CVD risk, but also hypoglycemia. As a result, guidelines relating to the therapeutic targets of glucose control have been under revision. Furthermore, some of the oral hypoglycemic therapies have come under scrutiny as they have been associated with increased CVD outcomes. As a result, all newer therapies have also been subject to intensive CVD evaluation to ensure CVD safety. Newer therapies targeting hyperglycemia with lower risks of hypoglycemia have also emerged.
This chapter will examine the role of glucose in the etiology and pathophysiology of CVD in diabetes. It will also explore some of the current controversies relating to plasma glucose and CVD risk and will describe the current guidance and evidence relating to the role of current glucose-lowering therapies and their effects on CVD risk.
Epidemiological Evidence for a Causal Role of Hyperglycemia in Cardiovascular Disease
Hyperglycemia as a risk factor for CVD has been established for many years. Mortality from CVD accounts for more than 60% of deaths in patients with type 2 diabetes mellitus and clearly accounts for this ultimate complication of diabetes [3, 8]. The association between differing degrees of hyperglycemia and CVD risk has been an area of debate. The United Kingdom Prospective Diabetes Study (UKPDS) demonstrated that the incidence of myocardial infarction rose by 14% per 1% rise in HBA1c [9]. This is in line with other studies showing that glucose is a continuous risk factor in people with both type 1 and type 2 diabetes.
Excess CVD risk is also observed in subjects with impaired glucose tolerance. The Bedford survey [10], the Whitehall study [11–13], and others [14, 15] have shown an increased mortality at the upper end of the blood glucose distribution before type 2 diabetes becomes apparent. Furthermore, differing associations have been observed along the spectrum of impaired fasting glycemia (IFG) and impaired glucose tolerance (IGT) in relation to CVD risk. In the Japanese Funagata Diabetes Study [16], survival analysis concluded that IGT and not IFG was associated with CVD outcome. Within this sample of approximately 2,500 subjects, IGT was associated with a doubling of CVD events relative to subjects with normoglycemia. The Diabetes Epidemiology: Collaborative Analysis of Diagnostic Criteria in Europe (DECODE) study jointly analyzed data from more than 10 prospective European cohort studies and included more than 22,000 subjects [17]. Death rates from all causes, cardiovascular and CHD, were higher in subjects with diabetes diagnosed by the two-hour postload plasma glucose than in those who did not meet these criteria. Significant increase in mortality was also observed in patients with IGT, whereas there was no difference in mortality between subjects with IFG and normal fasting glucose. In the recently published Emerging Risk Factor Collaboration study [3], a fasting plasma glucose greater than 5.6 mmol/L (100 mg/dL) was associated with increased mortality.
Other evidence supporting the role of hyperglycemia as a causative factor for CVD comes from studies where CVD risk has changed as a result of intensive treatment. As previously described in the UKPDS [18] of type 2 diabetes there was a 16% reduction in CVD (combined fatal or nonfatal MI and sudden death) in the intensive glycemic control arm (P = 0.052). In the Diabetes Control and Complications Trial (DCCT) there was a trend toward lower CVD risk with intensive control (risk reduction 41%), but the number of events was small [19]. In a nine-year post-DCCT study, follow-up of the cohort participants previously randomized to the intensive arm had a 42% reduction (P = 0.02) in CVD outcomes and a 57% reduction (P = 0.02) in the risk of nonfatal myocardial infarction, stroke, or CVD death compared with those in the standard arm [1].
While the above discussion demonstrates a causative association between hyperglycemia and CVD risk, debate also exists on the role of postprandial hyperglycemia versus fasting hyperglycemia to CVD risk. In Western society only a small amount of time is spent fasting and a far greater proportion of the day is spent in the postprandial state. Thus, it is not surprising that postprandial hyperglycemia is a greater contributor to blood glucose control than fasting glucose concentration [20]. The importance of the postprandial state also becomes apparent from the observation that therapies used to target postprandial glucose are more effective at lowering HBA1c than those aimed at lowering fasting glucose [21]. Several studies provide evidence supporting the suggestion that postprandial glucose levels are more closely associated with CVD risk than HBA1c or fasting glucose [22]. This has been observed in the DECODE study, described above [23], and by Bonora et al. [24]. In the latter, a cohort of 1,121 patients were followed for a mean of 52 months, with plasma glucose being assessed two hours after breakfast or lunch. It was observed that postprandial glucose was able to predict the onset of CVD, even after adjusting for other risk factors. Other studies such as the Hoorn study [25], the Honolulu Heart study [26], and the Chicago Heart study [27] have also shown that plasma glucose two hours after oral challenge is a powerful predictor of CVD risk.
To conclude, there is sufficient evidence to support the role of hyperglycemia in relation to CVD risk. Later in the chapter we will explore the effects of glucose variability and fluctuations on CVD risk and the underlying pathophysiological mechanisms by which these changes are brought about.
Glucose Fluctuations and Cardiovascular Risk
There is considerable evidence showing a link between postprandial hyperglycemia and cardiovascular risk [25, 27–29]. Furthermore, two-hour postprandial glucose is a better predictor of mortality in diabetes than is HBA1c [22, 25]. Later in this chapter, we will discuss the role that oxidative stress related to hyperglycemia plays in diabetes-related cardiovascular risk. There is also evidence that glucose fluctuations (the highs and lows) are associated with increased oxidative stress [30, 31]. Of interest is that postprandial glucose control with a short-acting insulin secretagogue (repaglinide v glibenclamide) is observed to result in regression of carotid artery intima-media thickness [32]. Similarly, the reduction of postmeal glucose excursions with a premeal bolus of a rapid insulin analog (aspart) is associated with a decrease in plasma markers of oxidative stress [33]. This provides direct evidence for a link with acute rather than chronic hyperglycemia. An increase in postprandial glucose is usually the major contributor of glucose variability; however, downward fluctuations are also important. For example, urinary markers of oxidative stress are associated with the mean amplitude of glycemic excursions [31]. Furthermore, in a study in which episodes of symptomatic angina were recorded in the presence of continuous glucose monitoring and a holter cardiac monitor, sudden changes in glucose levels (100 mg/dL/hr or 5.5 mmol/L/hr) were associated with more episodes of angina [34].
Important Physiological Mechanisms of Glucose-Mediated Cardiovascular Risk
In this section we will discuss the mechanisms of glucose-mediated CVD risk, focusing on the three areas of oxidative stress, autonomic dysfunction, and hypoglycemia.
Oxidative Stress, Advanced Glycosylation End Products, and Endothelial Dysfunction
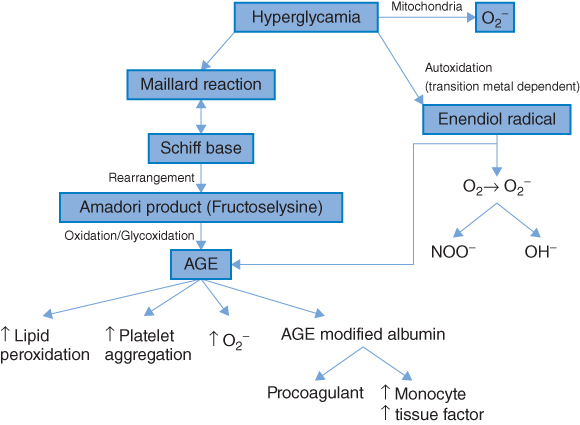
Considerable interest has developed in the role of free radical-mediated damage in many chronic disorders, in particular CVD, diabetes, and cancer [30, 35]. Free radicals are atoms or molecules that have one or more unpaired electrons in their atomic structure and are therefore highly reactive. Oxygen is the most ubiquitous of all biologically important chemical species and is a major source of reactive oxygen species (ROS). Increased oxidative stress results from an imbalance between oxidant production and antioxidant defenses [36]. Diabetes mellitus, obesity, micro- and macrovascular complications have been consistently associated with increased oxidative stress [37–39] and several studies have demonstrated that hyperglycemia per se is associated with increased oxidative stress [39, 40]. Furthermore, increased biomarkers of oxidative stress are independently associated with future CVD risk [41]. Possible mechanisms [36, 42] by which hyperglycemia may induce ROS formations are shown in Figure 5.1. As shown, hyperglycemia may result in the glucose-mediated nonenzymatic glycosylation of proteins (the Maillard reaction). The result of this is the formation of advanced glycosylation end products (AGEs). These not only increase ROS production but may also initiate a cascade of events which have harmful effects on the vascular system and are important in the etiology of both micro- and macrovascular complications relating to diabetes. Alternatively, glucose may undergo auto-oxidation to form a highly reactive enendiol radical. This may catalyze the conversion of molecular oxygen to O2.− (and hence increase ROS).
Figure 5.1 Causes and consequences of oxidative stress. ROS, Reactive oxygen species; MMP, Matrix metalloproteinases; VSMC, Vascular smooth muscle cell; HR, Hazard ratio. (Source: Stephens et al. 2009 [30]. Reproduced with permission of Elsevier.)
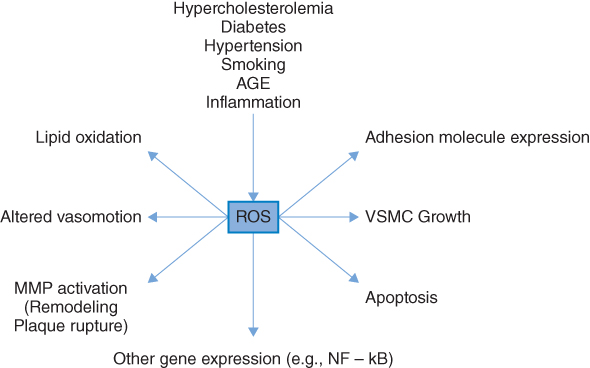
There is also an important direct mitochondrial mechanism that increases ROS [42] as a result of excess energy substrate (typically glucose and free fatty acids) entering into the citric acid cycle. The mitochondria of the endothelial cells of the vasculature may be particularly affected by “overfeeding,” as these cells are not dependent on insulin for glucose uptake and therefore take up glucose freely in the setting of hyperglycemia. As shown in Figure 5.2, increased OS is associated with many of the risk factors implicated in the pathophysiology of atherosclerosis [43]. All molecules are potential targets for ROS (proteins, lipids, and DNA), but because of their ubiquitous distribution within cell membranes, and their propensity to contain double bonds, unsaturated lipids are often targeted [42]. Increased oxidative stress is associated with the increased expression of cell adhesion molecules [44], important in the initiation of atherosclerosis [45]. Apart from the global effects associated with increased oxidative stress described above, more specific effects also occur. Low-density lipoprotein (LDL) is an important target of oxidation, and oxidative modification of LDL is a key step in the pathogenesis of atherosclerosis [46]. Elevated levels of oxidized LDL (Ox-LDL) are independently associated with increased atherosclerotic burden and increased CHD risk [47–49]. It is now clear that Ox-LDL, with its many oxidatively modified lipids and degradation products, contributes to the pathophysiology of both the initiation and progression of atherosclerosis.
Figure 5.2 Hyperglycemia-induced oxidative stress. AGE, Advanced glycosylation end products; O2., Superoxide radical; NOO., Peroxynitrite radical. (Source: Stephens et al. 2009 [30]. Reproduced with permission of Elsevier.)
Autonomic Dysfunction
Both cardiac and noncardiac autonomic neuropathy are risk factors for CVD in patients with diabetes [50]. These are associated with poor glycemic control and duration of diabetes [51, 52]. Of interest is that a hyperactive sympathetic system and a hypoactive parasympathetic system have been associated with CVD in subjects with diabetes [53]. Furthermore, in vitro studies suggest that hyperglycemia has an effect on postganglionic sympathetic neurons, resulting in functional changes in the perivascular sympathetic nerves due to changes in norepinephrine release [54]. This effect may partly be related to oxidative stress; however, hyperglycemia also has a direct inhibitory effect on ATP-sensitive potassium channels [55], which depolarize neuronal tissue and alter norepinephrine release [56]. There remains uncertainty on the underlying mechanisms relating to autonomic dysfunction, diabetes, and CVD risk. Recent studies suggest that adipokines such as leptin and tumour necrosis factor-alpha influence autonomic function [57]. Cardiac autonomic neuropathy is associated with multiple risk factors including duration of diabetes, severity of hyperglycemia, and the presence of coronary artery disease, but the precise contribution to CVD risk is difficult to ascertain. Nevertheless, recent analysis in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) study participants shows that cardiac autonomic neuropathy predicts all-cause and CVD mortality independently of baseline CVD, diabetes duration, and multiple other important CVD risk factors [42].
Hypoglycemia
Hypoglycemia is also associated with increased cardiovascular mortality [58, 59], although the mechanisms behind this remain unclear. A decrease in myocardial blood flow reserve during hypoglycemia may be a possible mechanism [60]. Alternatively, in vivo studies have demonstrated that acute hypoglycemia is associated with an increase in the concentration of a potent vasoconstrictor, Endothelin-1 [61]. As well as being associated with increased oxidative stress [62], hypoglycemia also has pro-inflammatory effects on the vasculature. These include increased neutrophil numbers and elevated neutrophil elastase [63]. These changes contribute to a hypercoagulable state associated with increased platelet aggregation and plasma concentrations of coagulation factors [29, 64]. Acute hypoglycemia has also been associated with long QT syndrome, which is associated with an increased risk of sudden cardiac death [65]. This may be related to sudden changes in catecholamine levels. Prolonged hypoglycemia may also have a direct detrimental effect on cardiac function because of the inability of the heart to utilize glucose, the preferred substrate instead of fatty acids (during acute myocardial ischemia), after exhaustion of myocardial glycogen reserves [66].
The Controversy of Intensive Glucose Management and Cardiovascular Risk
Intensive Glucose Control and Cardiovascular Mortality in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Study
Previously, several studies [67, 68] have clearly shown a direct relationship between HBA1c and CVD risk; however, the role of intensive glycemic control in reducing CVD events has not been well demonstrated. In the Diabetes Control and Complications Trial (DCCT) there was a trend toward a lower CVD risk with intensive control (risk reduction 41%), but the number of events was small [19]. In a nine-year post-DCCT study, follow-up of the cohort participants previously randomized to the intensive arm had a 42% reduction (P = 0.02) in CVD outcomes and a 57% reduction (P = 0.02) in the risk of nonfatal myocardial infarction, stroke, or CVD death compared with those in the standard arm [1]. In the UKPDS study [18], there was a 16% reduction in CVD (combined fatal or nonfatal MI and sudden death) in the intensive glycemic control arm (P = 0.052). However, further analysis showed a continuous association such that for every percentage point of lower median on-study HBA1c, there was a statistically significant 18% reduction in CVD events, again with no glycemic threshold.
In 2008, two studies (Table 5.1)—namely, the Action in Diabetes and Vascular Disease–Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) [69] and the Veterans Affairs Diabetes Trial (VADT) [70]—were published that showed no significant reduction in CVD outcomes with intensive glycemic control. Another trial, the Action to Control Cardiovascular Risk in Diabetes (ACCORD) [71], terminated prematurely due to the finding of increased mortality in participants randomized to intensive glycemic control with a target HBA1c <6%. The ACCORD trial was designed to examine the hypothesis that reducing blood glucose concentrations to near-normal levels in adults with type 2 diabetes at high risk of a CVD event would result in a reduction in nonfatal and fatal CVD. The participants were randomly assigned to receive therapy for intensive glycemia control (HBA1c <6.0%) or therapy for standard glycaemia control (HBA1c 7.0–7.9%). As stated, the ACCORD intensive glycemia control intervention was stopped early, after 3.5 years, because of higher mortality in this study arm (1.42% of patients died each year compared with 1.14% a year in the standard intervention arm; hazard ratio 1.22 [1.01–1.46]; P = 0.04) [71]. This increase in mortality was initially thought to be related to intensive-treatment-associated hypoglycemia. However, subsequent analysis of the ACCORD data suggests that this is not the case [72]. The later study showed that symptomatic hypoglycemia did not differ between the two groups and did not account for the difference in mortality between the two study arms.
Table 5.1 Comparison of ACCORD, ADVANCE, and VADT
| ACCORD | ADVANCE | VADT | |
| Number recruited | 10,251 | 11,140 | 1,791 |
| Median HBA1c (%) | 8.1 | 7.2 | 9.4 |
| HBA1c target (%) Intensive vs. standard | <6.0 vs. 7.0–7.9 | ≤6.5 vs. local guidelines | <6.0 (action if >6.5) vs. planned separation of 1.5 |
| Median follow-up (years) | 3.5 (terminated) | 5 | 5.6 |
| Median HBA1c (%) Intensive vs. standard | 6.4 vs. 7.5 | 6.3 vs. 7.0 | 6.9 vs. 8.5 |
| Primary outcome | Nonfatal MI, nonfatal stroke, CVD death | Microvascular plus macrovascular (nonfatal MI, nonfatal stroke, CVD death) | Nonfatal MI, nonfatal stroke, CVD death, hospitalization for heart failure, revascularization |
| HR for primary outcome | 0.90 (0.78–1.04) | 0.90 (0.82–0.98) macrovascular 0.94 (0.84–1.06) | 0.88 (0.74–1.05) |
| HR for mortality | 1.22 (1.01–1.46) | 0.93 (0.83–1.06) | 1.07 (0.81–1.42) |
| Reference | [71] | [69] | [70] |
Another study examined the ACCORD data to examine whether on-treatment HBA1c itself had an independent relationship with mortality [73]. This study found no evidence to suggest that lower average HBA1c was associated with higher mortality, which would have been the case if hypoglycemia had been the cause of death. Of interest is that a higher mortality rate was observed in both the intensive and standard therapy arms for individuals with a higher average and last HBA1c. This result was statistically significant in the intensive treatment arm only where a linear relationship was observed between HBA1c and mortality for an HBA1c between 6.0% and 9.0%.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


