Fig. 19.1
Panels (a) and (b): Demonstrating long QTc 0.54 s in an 18-month-old boy with sensorineural deafness. Panel (c): Marked T-wave alternans. Panel (d): Spontaneous ventricular fibrillation, which self-terminated. No genetic testing was available
Extensive molecular genetic studies in families and in isolated patients with LQTS have determined that mutations in at least 13 different genes may be responsible for causing this disorder (Table 19.1). Electrophysiologic and genetic characterization of LQTS patients has demonstrated that mutations in these different genes cause distinct manifestations of the LQTS, each with unique risks and different responses to therapy. The traditional concept that LQTS causes cardiac arrhythmias only at times of adrenaline excess has been replaced by a more precise view, in which the molecular subtype of LQTS strongly influences both the triggers for arrhythmia and the prognosis with treatment. However, as clinical genetic testing has become increasingly integrated into clinical care, a more complex understanding of the variability in clinical expression of LQTS has also emerged. Individuals with the same mutation can exhibit very different disease severity due to unknown background genetic influence, and up to 10 % of LQTS patients may carry multiple mutations in LQTS gene(s) [4].
Table 19.1
Genes in which mutations result in the long QT syndrome
Long QT locus | Gene | Gene function | Features |
|---|---|---|---|
LQT1 | KCNQ1 | Potassium channel; I(Ks) current | Sudden death with stress/exercise |
LQT2 | KCNH2 (HERG) | Potassium channel; I(Kr) current | Worse with hypokalemia |
LQT3 | SCN5A | Sodium channel | Sudden death during sleep; bradycardia |
LQT4 | ANK2 | Ankyrin-2; modifies sodium current | Sinus node dysfunction; atrial fibrillation |
LQT5 | KCNE1 | Potassium channel; I(Ks) current | Sudden death with stress/exercise |
LQT6 | KCNE2 | Potassium channel; I(Kr) current | |
LQT7 | KCNJ2 | Potassium channel; I(Kr) current | Andersen syndrome; periodic paralysis |
LQT8 | CACNA1C | L-type calcium channel | Timothy syndrome: syndactyly; 2:1 AV block; cognitive deficits |
LQT9 | CAV3 | Calveolin-3; modifies sodium current | Sudden death during sleep; bradycardia; SIDS |
LQT10 | SCNA4B | Sodium channel, beta subunit | Bradycardia; 2:1 AV block |
LQT11 | AKAP9 | Modifies KCNQ1 function | |
LQT12 | SNTA1 | Syntrophin; modifies sodium current | |
LQT13 | KCNJ5 | Potassium channel; I(Kr) current | Atrial fibrillation |
I Ks Defects
Defects of KCNQ1 and KCNE1, the alpha and beta subunits of the potassium channel responsible for the I Ks current (Fig. 19.2), cause LQT1 and LQT5, respectively. Mutation of a single copy of either of these genes may cause autosomal dominant LQTS. However, mutations in both copies of one of these genes can result in severely symptomatic LQTS that is characterized by sensorineural deafness, as initially described by Jervell and Lange-Nielsen. Recently, a mutation of AKAP9, which encodes for part of a protein complex that regulates KCNQ1 function, has also been demonstrated to result in LQTS, now designated as LQT11. The I Ks current is responsible for the slowly activating outward potassium current that contributes to the return of the myocyte to its resting potential following a depolarization (Chap. 2). LQTS-causing mutations may occur anywhere in the genes that encode these two channels, but the more severe defects appear to be those that result in the production of an ion channel subunit with abnormal potassium conduction properties. These abnormal subunits can complex with normal subunits so that the vast majority of I Ks channels within the myocyte are either nonfunctional or function abnormally. Mutations that do not produce an abnormal protein product result in approximately half the number of fully functional I Ks channels, causing less severe manifestation of the LQTS. With some of the mutations, no abnormalities are noted when a patient inherits only one defective copy of the gene and the LQTS only becomes apparent when two altered copies of the gene are inherited as is noted in the Jervell and Lange-Nielsen syndrome. However, in general, the location of the mutation within the gene has not been demonstrated to reliably predict prognosis. This analysis has been most thoroughly examined in LQT1 [5]. An exception is the C-loop of KCNQ1 which is associated with a high rate of cardiac events in LQT1 [6].
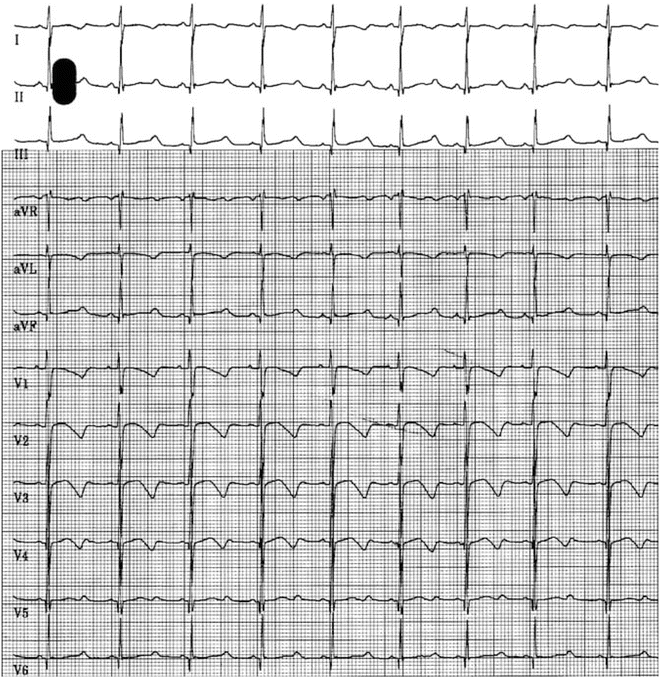
Fig. 19.2
12 Lead ECG in a 8-year-old boy with sudden loss of consciousness for 10 s. Note the QTc of 0.48 s in Lead 2 and V5
I Kr Defects
Defects of KCNH2 and KCNE2, the alpha and beta subunits of the potassium channel responsible for the rapid potassium (I Kr) current, cause LQT2 and LQT6, respectively. Mutations in these genes have primarily been associated with autosomal dominant LQTS. I Kr, the current responsible for the initiation of cardiac repolarization following excitation-contraction, has some unique properties that affect the clinical manifestations of the LQTS in these patients. The potassium conductivity of the I Kr channel is very sensitive to extracellular concentrations of potassium [7]. Independent of the transmembrane electrochemical gradient, which, in large part, is maintained by the difference between the intracellular and extracellular potassium concentrations, the capacity of the channel to conduct an outward potassium current (i.e., I Kr) varies directly with the extracellular potassium concentration. As the extracellular potassium rises, the outward potassium current through the channel increases, and conversely, a low extracellular potassium concentration inhibits the channel’s function. Therefore, patients that have KCNH2 or KCNE2 mutations that inhibit the function of the I Kr channel are particularly sensitive to low extracellular potassium concentrations that further reduce the conductance of the channel.
Mutations involving other inward rectifier potassium channels have recently been determined to cause a long QT phenotype and ventricular arrhythmias. Some mutations of KCNJ2, which has been implicated as the gene responsible for Andersen syndrome (see below) and encodes the channel responsible for the Kir2.1 current, have been determined to cause significant QT prolongation. A mutation of KCNJ5, which encodes for a G protein-regulated inward rectifier potassium channel, was detected in a 4-generation family with LQTS. The mutation showed incomplete penetrance within the family suggesting the presence of disease modifiers in determining disease expression [8].
Mutations Affecting Sodium Conductance
Patients with defects of the cardiac sodium channel, SCN5A, which account for less than 10 % of the reported LQTS cases, are more susceptible to arrhythmias and sudden death during times of bradycardia and may die in their sleep. In general, they appear less likely to have an arrhythmia than patients with potassium channel defects but their arrhythmias are more likely to be fatal since they are often not observed. The LQTS-causing SCN5A defects described to date appear to impair the ability of the channel to completely inactivate after depolarization, allowing persistent inward leak of Na+ ions. This may lead to prolongation of the plateau phase of cardiac depolarization, thereby delaying the onset of repolarization and resulting in excess calcium loading of the myocyte. The increased intracellular calcium may allow the development of early after-depolarizations and the generation of ventricular arrhythmias, particularly at slower heart rates. Interestingly, only a subset of SCN5A mutations is capable of causing LQTS. As opposed to the gain-of-function type of mutations described above for SCN5A-associated LQTS, other mutations in SCN5A result in inhibition of inward sodium current or result in a nonfunctional channel [9]. These mutations have been associated with a wide range of distinct clinical phenotypes, including Brugada syndrome, idiopathic ventricular fibrillation, FAF, sick sinus syndrome, and cardiac conduction defects.
More recently, mutations have been identified in proteins that regulate the function of the SCN5A channel, causing QT prolongation, ventricular arrhythmias, and sudden death. Mutations have been identified in SCN4B (the beta subunit of the sodium channel), syntrophin and ankyrin-2 (membrane-bound cytoskeletal proteins), and CAV3 (a membrane protein that regulates the formation and recycling of specialized membrane compartments). In each case, the LQTS-causing mutation results in enhanced late sodium current and leads to sinus bradycardia, ventricular arrhythmias, and sudden death, predominantly during sleep. In addition, since these proteins link the cellular cytoskeleton to membrane-bound multi-protein complexes, mutations in these proteins can result in a spectrum of disorders, including cardiomyopathies (SCN5A, CAV3) and skeletal muscular dystrophies (CAV3).
Calcium Handling Abnormalities
To date, only one of the subtypes of LQTS has been determined to be due to a primary abnormality of calcium handling. Mutations of CACNA1C, the alpha subunit of the L-type calcium channel cause Timothy Syndrome, which is characterized by lethal ventricular arrhythmias, severe QT prolongation, webbing, or syndactyly of the fingers and toes, and cognitive deficits [10]. Not unlike other LQTS-causing genes, mutations of CACNA1C can result in a range of phenotypes depending on the domain affected and the nature of the amino acid substitution. As with SCN5A, some mutations can result in a Brugada syndrome phenotype.
Drug-Induced LQTS
It has long been known that some patients with normal QT intervals could have marked QT lengthening and torsades de pointes in response to certain medications (for more complete list, see http://www.sads.org/ and http://crediblemeds.org/pdftemp/pdf/US-DrugsToAvoidList.pdf). These medications typically interact with function of the potassium channel encoded by the KCNH2 gene, associated with congenital LQT2. These apparently “idiosyncratic” reactions have now been attributed in many cases to inherited, subclinical LQTS mutations that only become clinically apparent when there is another compromise to cardiac repolarization as occurs with certain medications that block sodium or potassium channels. The effect of these medicines on the cardiac repolarization may be particularly dramatic when they are combined with other agents such as that delay their metabolic degradation by inhibiting the P450 system or when they occur in a patient with hypokalemia, an additional inhibitor of the I Kr current.
Diagnosis of LQTS
The diagnosis of the LQTS is based on the measurement of the QT interval on the 12-lead electrocardiogram and corrected for heart rate using Bazett’s formula [QT/(R − R)1/2]. Corrected QT intervals of 0.47 in males and 0.48 in females were the initial threshold values for identifying patients with LQTS. However, as genetic testing has become increasingly utilized, a major clinical challenge has emerged. Up to 30 % of individuals with known LQT-associated mutations identified through family screening may have normal QT intervals. This observation is likely due to the presence of multiple other influences on the QT interval that are yet to be defined. Moreover, the QT interval in the normal population exhibits a broad distribution. As a consequence, the QT interval thresholds above will have a low sensitivity for diagnosis of LQTS in individuals with clearly positive family histories for the disorder. On the contrary, lower thresholds would have exceedingly high false positive rates if applied to the general population. A scoring system was devised to improve the sensitivity and specificity of the diagnostic criteria for the LQTS. The scoring system categorizes each patient as high or low risk for having clinical LQTS based on their symptoms, their family history, their measured corrected QT interval and their T wave morphology [11, 12] (Table 19.2). While this appears to be a significant improvement in establishing the clinical diagnosis of LQTS, it has been developed using the most common types of LQTS, LQT1, and LQT2, as a model and may be less helpful for the less common subtypes.
Table 19.2
Long QT syndrome (LQTS) diagnostic criteria
Points | |
|---|---|
Electrocardiographic findings a | |
QTcb | |
≥ 480 ms | 3 |
460–479 ms | 2 |
450–459 ms (males only) | 1 |
QTcb at the fourth minute of recovery from an exercise stress test ≥ 480 ms | 1 |
Torsades des pointesc | 2 |
T wave alternans | 1 |
Notched T wave in 3 leads | 1 |
Heart rate less than the second percentile for age | 0.5 |
Clinical history | |
Syncopec | |
With stress | 2 |
Without stress | 1 |
Congenital deafness | 0.5 |
Family history | |
Family members with diagnosed LQTS | 1 |
Unexplained sudden cardiac death under the age of 50 years in immediate family member (not diagnosed with known or suspected LQTS) | 0.5 |
Probability of LQTS | Total score |
Low | ≤1 |
Intermediate | 1.5–3.0 |
High | ≥3.5 |
When available, genetic testing is often the best screening approach for individuals with a family history of LQTS, provided that a mutation is identified in the family member (see below). Genetic testing has, however, presented a treatment dilemma in the form of the genotype positive/phenotype negative patient. These are patients who have been identified as carriers of the LQTS (they have one copy of a defective LQTS gene), but they have never had symptoms and their electrocardiogram appears entirely normal. These patients and all first-degree relatives of patients with LQTS who have not had genetic testing should avoid medications on the list of those known to prolong the QT interval (http://www.sads.org/; www.qtdrugs.org).
Beyond careful ECG QT measurements, genetic testing and the clinical scoring system described above, adjunctive testing has proven to be of little benefit. Noninvasive electrophysiologic testing may have a role in diagnosis or prognosis of LQTS. Provocative testing with epinephrine infusion has been used to diagnose LQTS not evident on a resting electrocardiogram. However, this test should be used with caution, since the false positive rate is unknown. Holter monitoring may be of benefit to determine QT intervals at various times during the day, since the QT interval at one single moment in time may not always be reflective of an individual’s arrhythmic risk. In addition, exercise ECG may be helpful in determining a lack of rate adaptation of the QT interval, which occurs specifically in LQT1. Thus, a negative exercise ECG test would not exclude other forms of LQTS. In addition, measurements of the QT interval should be focused during early exercise, before the heart rate exceeds the threshold of accurate estimation with the Bazett calculation.
Treatment of LQTS
All patients with LQTS should avoid competitive athletics and all medications known to prolong the QT interval (as well as any alternative therapies that have unknown effects on the QT interval). Further recommendations and treatment choices are tailored to the individual patient. For each patient, the clinical presentation and genetic diagnosis significantly impact risk and therapeutic options in LQTS. The molecular genetic diagnosis is now an efficient starting point for clinical decision making in LQTS and is endorsed by the Heart Rhythm Society/European Heart Rhythm Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies [13]. Once a specific mutation is identified, a treatment plan (which may include pharmacotherapy, device therapy, and/or activity recommendations) can be refined based on the characteristics of the identified subtype of LQTS.
Patients with I Ks defects [who have mutations of KCNQ1 (LQT1), KCNE1 (LQT5), and potentially AKAP9 (LQT11)] are at greatest risk of arrhythmias and sudden death during adrenergic surges that occur during fright or anger, the “flight or fight response.” Beta blockade treatment has now been well proven to improve survival in LQT1, which represents the largest genetic subgroup of patients with LQTS. It may follow that patients with LQT5, also characterized by a reduction in I Ks current, may derive a similar benefit from beta blocker therapy, though clinical data on this much smaller subgroup has not yet verified. Previous studies have demonstrated that LQT1 patients who are strictly compliant to adequately dosed beta blocker therapy generally have a good prognosis. The choice of beta blocker is important as treatment with either nadolol or propranolol is superior to metoprolol [14]. All patients with LQTS are recommended to avoid competitive athletics, though the greatest risk of adrenergic-mediated events is in LQT1. Interestingly, studies have also demonstrated that patients with KCNQ1 defects, and presumably with KCNE1 defects as well, are particularly susceptible to arrhythmias while swimming. This observation has led to the recommendation that all patients with the LQTS should not swim alone or without flotation gear. Additional studies may indicate that only those patients with I Ks defects need to avoid swimming.
Patients with I Kr defects [who have mutations of KCNH2 (LQT2) and KCNE2 (LQT6)] are uniquely predisposed to arrhythmias triggered by sudden loud noises and should remove loud alarm clocks and bedside phones. Postpartum arrhythmias are also particularly common in LQT2, likely worsened in the setting of sleep deprivation and stress, and these women should be counseled appropriately. Patients with LQT2-type LQTS have an intermediate response to beta blockade therapy so alternative and supplementary therapies have been investigated.
Hypokalemia should be avoided, and dietary intake of potassium-rich foods would theoretically be of benefit for LQT2 since extracellular potassium impacts the function of KCNH2 channels. Some clinical data has supported the use of potassium supplements or spironolactone to shorten the QT interval in LQT2, though no trial data has demonstrated a reduction in cardiac events.
LQT3, which is due to sodium channel mutations, follows a distinct clinical course from the more common LQTS subtypes which are due to abnormalities of potassium conductance (Figs. 19.3 and 19.4). Ventricular arrhythmias in LQT3 patients are unpredictable and often occur during sleep. Beta blockers have shown a lower success rate in LQT3, although newer data, primarily from a mouse model of LQT3, suggests there may be some benefit, particularly with propranolol which has a sodium channel blocking effect not demonstrated by other beta blockers [15]. The incomplete protection provided by beta blockers has prompted study of specific sodium channel blockade in LQT3. Mexiletine showed early promise as a therapy for LQT3 based on cellular models, and on observations in a subset of patients [16]. However, a more complex view has emerged, since some SCN5A mutations appear to cause an overlap syndrome with features of both LQT3 and Brugada. In these patients, treatment with mexiletine may not improve the sodium channel defect and may even be harmful. Therefore, further study will be required to determine if mexiletine is appropriate for a subset of LQT3 patients. Cardiac pacing has been suggested to have a potential benefit in preventing bradycardia-associated events in LQT3 but has not been prospectively studied.
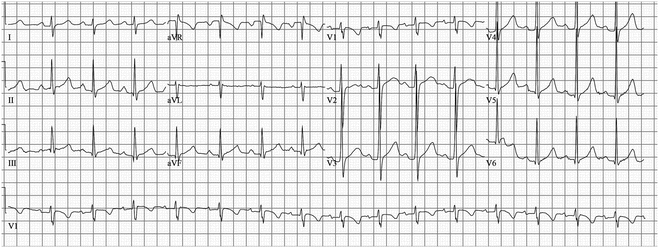
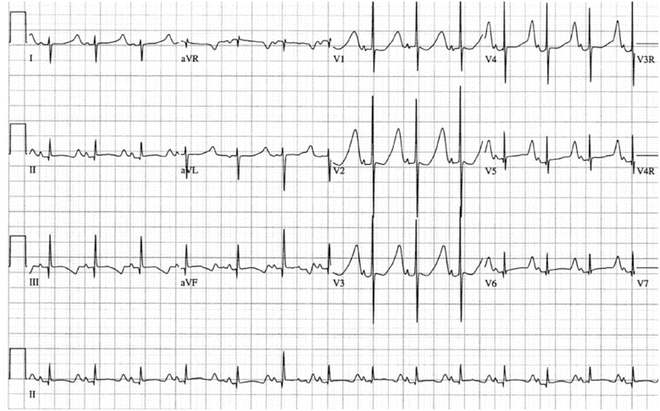
Fig. 19.3
7-Year-old boy with frequent syncope episodes; he was genotype positive for LQT3. He has a normal QTc
Fig. 19.4
12-Lead electrocardiogram in 2-day-old infant. Note the long flat ST segment and relatively symmetrical T-wave typical of LQTS3 and in contrast to the ECG phenotype in Fig. 19.3
LQTS is largely a medically treated disease. However, a minority of patients will have a high enough risk to warrant an implantable cardioverter-defibrillator (ICD), and, in these patients, an ICD can be life-saving. Patients who continue to be symptomatic on therapy, have had a cardiac arrest, or have marked QT prolongation (>500 ms) are potential candidates for an ICD.
Brugada Syndrome
The Brugada syndrome (BS) is characterized by ST segment elevation in the right precordial leads (V1–V3) unrelated to other factors such as ischemia, electrolyte abnormalities, or structural cardiopulmonary disease [17] (Fig. 19.5). Patients with Brugada syndrome are predisposed to sudden cardiac death due to polymorphic ventricular tachycardia that degenerates to ventricular fibrillation. It is inherited as an autosomal dominant disorder with an incidence of 5–66 per 10,000 individuals. There is a much higher incidence occurring in areas of Southeast Asia, and a male predominance (8:1). The symptoms, typically syncope or cardiac arrest, usually do not appear until the third to fourth decade of life.
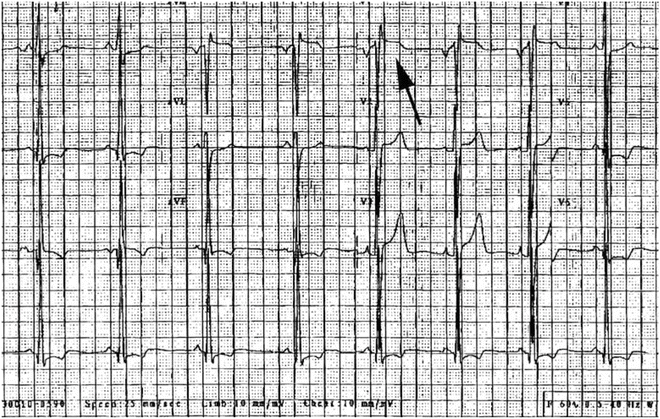
Fig. 19.5
Elevation of ST segment in V1 (arrow) in a 15-year-old boy with family history of Brugada syndrome
Genetic studies have determined that more than one gene is capable of causing the Brugada syndrome. The first gene identified as responsible for causing BS in some patients was the SCN5A cardiac sodium channel. Unlike SCN5A mutations that cause LQTS, BS-causing SCN5A mutations reduce the sodium channel current, either through a reduction in the number of channels and/or the diminished function of nearly half of the channels. Many of these mutations introduce a premature truncation codon, resulting in an unstable mRNA transcript and a consequent shortage of the protein product. However, the distinction electrophysiologically between Brugada syndrome and LQT3 may not always be clear, since an overlap syndrome containing features of both may arise from certain mutations, as discussed above. The diminished inward sodium current in Brugada syndrome due to SCN5A mutations is thought to allow a prominent transient outward potassium current (Ito) in the right ventricular epicardium, leading to an epicardial to endocardial voltage gradient. This produces the ST elevation that is characteristic of the disorder seen prominently in the right ventricular ECG leads (V1–V2) and may lead to re-excitation (phase 2 re-entry) in the ventricular epicardium, triggering ventricular arrhythmias.
BS-causing mutations have also been identified in GPD1L (a presumed phosphate dehydrogenase that regulates the sodium current), CACNA1C (the alpha subunit of the L-type calcium channel), SCN1B (a beta subunit of the cardiac sodium channel), SCN3B (a beta subunit of the cardiac sodium channel), KCNE3 (a beta subunit of the KCNQ1 potassium channel), and HCN4 (a hyperpolarization-activated and cyclic-nucleotide-gated cation channel that controls pacemaker activity in the sinus node). Collectively, these remain very rare causes of BS and only SCN5A, GPD1L, and CACNA1C mutations have been noted in more than a single family.
Diagnosis
Like LQTS, the diagnosis of BS has relied upon the characteristics of the 12 lead electrocardiogram, but also with less than optimal sensitivity and specificity. Three distinct right precordial ST wave morphologies have been described (Fig. 19.6) In Type 1 BS, there is prominent “coved” J point peak with an ST elevation of >0.2 mV followed by a negative T wave with essentially no intervening isoelectric phase. In Type II, there is a prominent J point peak (>0.2 mV) followed by a positive or biphasic T wave resulting in a saddleback appearance. In Type III, the J point elevation is less pronounced (>0.1 mV) and may be of either the “cove” or “saddleback” type. The ECG abnormalities may mimic RBBB, especially in patients with Type I BS. The BS pattern can be differentiated from RBBB by the absence of a wide S wave in lead I and in the left lateral precordial leads.
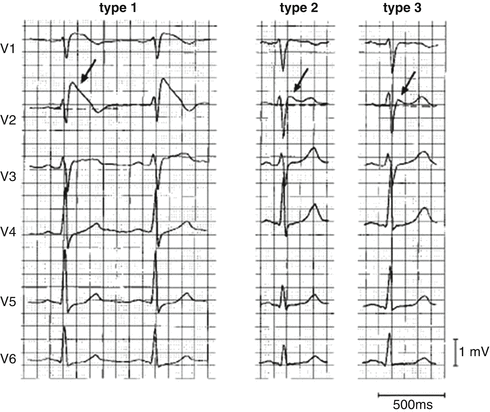
Fig. 19.6
Three types of QRS morphology in the Brugada syndrome (Reprinted with permission from Wilde AA, et.al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002;106(19): 2514–9. With permission from Wolters Kluwer Health)
The typical ECG appearance may not always be present but may be unmasked by intravenous infusion of sodium channel blockers such as ajmaline (1 mg/kg at 10 mg/min), flecainide [2 mg/kg (maximum 150 mg) over 10 min], or procainamide (10–15 mg/kg; 100 mg/min, the only medication FDA approved in the United States). However, sensitivity and specificity of the provocative testing has not been established and, in a patient that does have BS, may precipitate significant ventricular arrhythmias including ventricular fibrillation. Furthermore, there is some evidence to suggest that asymptomatic patients, who only demonstrate ECG changes with provocative testing, may have a benign course [18].
Genetic testing will have an increasingly important role in helping to understand the clinical spectrum of BS. Current genetic testing formats may only detect a putative mutation in approximately 20–30 % of the BS cases but exome-wide sequencing, which is being increasingly utilized in the clinical setting, may reveal additional cases of the rare genetic causes and identify novel genes, especially when performed in families with more than one affected individual. In the future, as more BS genes are identified and their manifestations more uniquely characterized, genetic testing may help make the diagnosis and guide the course of therapy.
Management
Treatment is required for all symptomatic BS patients. Unfortunately, pharmacotherapy has not proven to be beneficial. There is some theoretical argument for the use of quinidine to treat this disorder since quinidine, at low doses, is a potent inhibitor of the Ito potassium current. However, quinidine has not been prospectively studied in BS patients. Interestingly, there is also data to suggest that mexiletine, a sodium channel blocker, may improve trafficking of the mutant protein, allowing it to be transported correctly to the cell membrane where its function can help improve sodium conductance into the myocyte. However, the only treatment currently demonstrated to be effective in reducing mortality in BS patients is placement of an ICD. While ICD placement is clearly indicated in all BS patients with symptoms of syncope, there is controversy concerning their use in asymptomatic BS patients, particularly children and young adults. When an individual is identified during the screening of a family with symptomatic Brugada syndrome, it could be argued that ICD placement should be considered. Approximately 30 % of asymptomatic individuals will have a ventricular arrhythmia within 3 years of evaluation, the same as the percentage of symptomatic individuals that will experience a recurrent ventricular arrhythmia within 3 years. The only major risk factor identified in asymptomatic patients is a spontaneous type I ECG pattern, which confers an intermediate risk of ventricular arrhythmias. A 24-h Holter monitor may be useful in determining whether a spontaneous type I pattern emerges at times of higher vagal tone in these patients, such as after a large meal or during the night. While provocative drug challenge is useful diagnostically, it does not identify a higher risk group with Brugada syndrome. Electrophysiologic testing was initially shown to identify patients with inducible ventricular fibrillation who appeared to have a higher risk of clinical events. However, subsequent studies have questioned whether inducible VF is a reliable marker of events in Brugada syndrome.
Short QT Syndrome
While the dangers of prolongation of the QT interval have been recognized since the LQTS was first described in the late 1950s and early 1960s, only recently has it been discovered that a very short QT interval may also predispose to life-threatening arrhythmias and sudden death [19]. The short QT syndrome is very rare in comparison to LQTS and is characterized by a QTc (Bazett’s formula) less than 320 ms (Fig. 19.7). It is frequently associated with atrial fibrillation to the extent that the diagnosis should be strongly considered in young patients with isolated atrial fibrillation [20]. Establishing the diagnosis is vital since patients with the disorder have a high incidence of syncope and sudden death due to ventricular tachyarrhythmias. As with the LQTS, the short QT syndrome is a heterogeneous disorder with mutations in at least three different genes capable of causing the clinical manifestations. The mutations described to date are all gain-of-function mutations in genes in which loss of function can lead to the long QT or Anderson syndromes: KCNH2 (I Kr), KCNQ1 (I Ks), and KCNJ2 (I Kt). Treatment guidelines are still being established but case reports have recommended the use of ICD devices. Furthermore, quinidine may be of benefit, as adjunctive therapy in those patients with an ICD or primary therapy in those without, at least in patients with short QT syndrome due to mutations in the KCNH2 (HERG) channel.

Fig. 19.7
ECG from 14-year-old boy with aborted ventricular fibrillation. Note the QT 0.28 s
Early Repolarization-Associated Ventricular Arrhythmias
Early repolarization was long known as a benign clinical entity until the last decade. Several isolated reports, followed by population studies and case series have now suggested an increased risk of ventricular arrhythmias related to early repolarization, though this risk appears to be very modest on a population level. In the largest case series of patients with sudden cardiac arrest of unknown cause (n = 206), early repolarization, defined as a J point elevation regardless of ST elevation, was much more common than in controls (31 % vs. 5 %, p < 0.001) [21]. Subsequent to this report, there has been an argument that the key ECG feature potentially associated with ventricular arrhythmias is the J point elevation with a subsequent horizontal ST segment, as opposed to the ascending ST elevation frequently seen in the young, especially in male athletes, which is likely benign [22]. Other population studies have countered these findings, with no evidence of an increased risk of sudden death, regardless of the type of J point or ST segment elevation [23]. While the genetic underpinnings of early repolarization are not yet understood, Gourraud et al. published a series of 22 sudden deaths among 4 families associated with early repolarization. They did extensive screening and evaluation of these families (171 screened relatives), and their findings suggest that early repolarization in these families is inherited as an autosomal dominant trait with an increased risk of sudden death [24]. Therefore, while the vast majority of individuals with an early repolarization pattern on ECG, regardless of the morphology of the pattern, will not suffer from life-threatening arrhythmias, there may be selected individuals and families that require closer monitoring. A family history of early sudden cardiac death and syncope suspicious for arrhythmia may warrant closer scrutiny for the possibility of familial early repolarization patterns. More precise characterization of the clinical features that could distinguish those at risk from the large number of individuals with the common, apparently benign, variant of early repolarization will be a major challenge.
Familial Ventricular Fibrillation
Several families have been described with familial ventricular fibrillation (FVF). In one instance, the disorder was attributed to a mutation of the SCN5A gene but the patients did not demonstrate the electrocardiographic findings of Brugada syndrome. Additional FVF genes likely remain to be identified since not all patients with the disorder have been demonstrated to have SCN5A defects. A second locus was definitively linked to chromosome 7q36.2 in a Dutch cohort. A noncoding mutation was noted in the promoter region for the DPP6 gene which encodes for a component of the transient outward current in cardiac myocytes.
Catecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia has been demonstrated to be due to abnormal calcium release from the sarcoplasmic reticulum. It is a relatively rare heritable disorder that is characterized by adrenergic-induced ventricular tachyarrhythmia and sudden death (see Fig. 19.8). CVPT has been determined to result most commonly from an autosomal dominant mutation in the cardiac ryanodine receptor type 2 (RYR2), the channel that releases calcium from sarcoplasmic reticulum (SR) stores, or an autosomal recessive mutation (or compound heterozygous mutations) in calsequestrin, a molecule that helps to sequester calcium in the SR and interacts with ryanodine. More recently, CPVT-causing mutations have also been identified in CALM1 (which encodes for calmodulin, a key calcium-binding protein involved in intracellular calcium regulation), and TRDN (which encodes for triadin, a regulator of calcium release that links calsequestrin to the ryanodine receptor). Therefore, CPVT is primarily attributable to disordered calcium regulation.
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
