The Human Genome
The term genome refers to the entire chromosomal DNA including the genes responsible for an organism. The proteome refers to all of the proteins responsible for an organism. Genes exert their influence directly through producing proteins or indirectly through regulating protein-coding genes. Each gene produces a unique protein, referred to as a polypeptide. Some proteins are made of two or more polypeptides and a significant proportion of genes use alternative splicing to produce more than one form of the same polypeptide. In addition to genes that encode proteins, there are genes that transcribe RNA without encoding protein. The human genome within each cell consists of approximately 3 billion bases in the form of 23 pairs of chromosomes of which 22 pairs are homologous (their sequences are similar) referred to as autosomes, and the remaining pair contain the sex chromosomes which, in the male, consists of X and Y and, in the female, two X chromosomes. Each chromosome is a long molecule made of DNA. DNA is comprised of only four bases: adenine (A), guanine (G), cytosine (C), and thymidine (T) (see Chap. 6). The sequence of these four bases determines all inherited characteristics. The average length of a chromosome is about 135,000,000 base pairs (bp). The longest chromosome, chromosome 1, has more than 250,000,000 bp. The smallest, chromosome 21, has only 50,000,000 bp. The 23 chromosomes together contain a total of 3 billion bp (Table 10–1). The chromosomes contain the genes which themselves are discrete units with a start and stop point and vary in size from 10,000 to more than 2,000,000 bp. The estimated average size of a gene is about 20,000 bp. Most genes encode RNA transcripts which exert their ultimate influence through regulating protein-coding genes but themselves do not code for a messenger RNA (mRNA) or protein. Only a minority of RNA transcripts encode proteins. The genes are nonrandomly distributed along chromosome into gene-rich and gene-poor regions. The ends of chromosome are referred to as telomeres. The centromeres are those regions of chromosome that attach to the mitotic apparatus during cell division. Both telomeres and centromeres are comparatively gene-poor regions. Embedded in the DNA sequence of each chromosome are genes that encode for proteins (2% of the DNA)1 and others which encode for all of the RNAs. Interspersed between the sequences coding for proteins and RNAs are sequences to which factors bind to regulate when and how much of a gene is transcribed into protein or RNA. These sequences are referred as regulatory DNA elements. The factors binding to these elements are proteins referred to as transcription factors. These proteins have specific DNA-binding sites enabling them to attach to the regulatory DNA element to exert their control over transcription.
It is now postulated that most of genomic DNA is transcribed into RNA transcripts with the end product being primarily RNA and the remainder proteins. Approximately 10% of DNA sequence is transcribed into mRNA, of which 2% exits the nucleus to serve as templates for proteins. It is estimated that 46% of the human genome is composed of DNA from mobile elements transposed into the human genome over the past 150 to 200 million years.2 It is estimated that the human genome contains 23,000 genes with each encoding for a single polypeptide; but through alternative splicing of the exons, each gene may produce several slightly modified forms of that polypeptide. Thus, it is likely that despite the 23,000 genes, we may have over 100,000 proteins. The DNA is bound to a series of support proteins belonging to the histone family2 which maintains the structure of the chromosome. In addition to their support role, histones play a major function in the epigenetic regulation3 of gene expression (see Chap. 6).
Chromosomal Loci, Genes, and Alleles
The precise position of each gene on the chromosome is identical from person to person and is referred to as the chromosomal locus. Chromosome loci are designated by giving the chromosome number (1–23, X, or Y) and; whether it is on the long (q) or short arm (p) and the subband region. Subregions reflect the banding patterns of chromosomes based on Geimsa staining patterns. An example would be the chromosomal locus for the gene that encodes for angiotensin-converting enzyme (ACE) is designated 17q23. Each pair of autosomal homologous chromosomes carry the same set of genes with one inherited from each parent. Despite their homology and the overall function of the genes being identical between individuals, nearly all genes have considerable genetic variation. This variation extends to homologous chromosomes, where differences in sequence can be found between most genes when considering the maternally versus paternally inherited chromosomes. These differences are referred to as alleles. An example of this is the gene encoding for ACE located at 17q23 is present at this locus on each homologous chromosome from both parents. However, the ACE gene is known to exist in the general population in three forms referred to as alleles designated D, DI, and II, where D refers to a deletion of 250 bp within the ACE gene. The particular allele accompanying the 17q23 locus will depend on which alleles are inherited from the parents. While both genes or alleles encode for ACE and convert angiotensinogen to angiotensin II, there is increased plasma enzyme activity associated with the D form and studies suggest homozygosity for the D gene (DD), predisposes to cardiac hypertrophy.4,5 Alleles can refer to single bp difference, insertions, or deletions. Most genes exhibit multiple alleles.
The Human Genome Sequence—Blueprint for Human Life
The Human Genome Project had as its goals to map and sequence the entire DNA of the human genome. The Human Genome Project was the first large international effort in the history of biological research.6 The Human Genome Project was initiated on October 1, 1990; a rough draft of 90% of the DNA sequences was available to the public in 20006 and the complete sequence was available in 2003.7 The sequence of each gene was entered into a publicly accessible database and is freely available. In the United States, GenBank (available at ) run by the National Center for Biotechnology Information, serves as the public repository of sequence information.
The Human Genome Project contains the blueprint for the development of a single fertilized egg into a complex organism of more than 1013 cells. In addition to completing the complete sequence, other goals were also achieved. Genetic maps were established where variable sequences among individuals were placed in reference to each other, and then ultimately to the genome sequence itself. This goal was completed with thousands of markers spaced less than one million base pairs apart spanning the entire human genome.8 The genetic map was the necessary tool for widespread application of genetic linkage analysis, a technique which has led to the mapping of numerous loci and genes responsible for single-gene disorders including several diseases of the cardiovascular system (see Chap. 81). It is estimated there are about 6000 rare inherited single-gene disorders of which more than 2000 genes have been identified.9,10 The catalogue of single-gene disorders is found in the Online Mendelian Inheritance in Man, also known as OMIM, . OMIM documents all single-gene disorders including disease gene information along with correlates of mutations and phenotypes. Single-gene disorders, often referred to as Mendelian disorders, are those where mutations in a single gene produce the trait of interest. A number of important cardiovascular phenotypes are produced by mutations in single genes, for example, hypertrophic cardiomyopathy and Marfan syndrome (see Chap. 81). Most of these rare cardiovascular traits are dominantly inherited. The availability of genetic maps and the human genome sequence greatly facilitates establishing novel associations between genes and phenotypes. For example, rare dominant cardiovascular traits can be mapped with as few as seven affected individuals in a multigeneration family. For recessive traits, mapping and gene identification can be achieved with even fewer individuals, especially if consanguinity is present. Single families can prove highly valuable for mapping and identifying genes, emphasizing the importance of the astute clinician in cardiovascular genetics.
A genetic map of the human genome designates the locus of a gene as previously indicated by the example of the ACE gene being located at 17q23. This simply indicates the gene is on chromosome 17. If one were to compare this to a postal address, the chromosome number is the country, the q or p arm is the city, and the subband number lacks precise molecular specificity but does refine the genetic region. The resolution is such that the gene will be within a few million base pairs along with many other genes. It is now possible with the physical map to precisely locate the gene by its sequence in the human genome. This is equivalent to the street address. Concomitant with the sequencing of the human genome was the integration of the chromosomal signpost sequences spanning the genome that are expressed as mRNA. These tags identified throughout the genome are referred to as expressed sequenced tags (ESTs).7 ESTs consist of sequences of 200 to 300 bps which are unique and represent a specific gene. To be unique, ESTs were preferentially defined from the 3′ untranslated regions of genes that tend to have less homology to related family members. Not all ESTs are derived from 3′ untranslated regions, but these regions are more highly represented in the EST databases. ESTs were integrated into the human genome sequence and have been invaluable in assisting in the pursuit of human genes. The sequence of each EST in GenBank has been cloned and stored in bacteria referred to as a library of human ESTs. Most of the ESTs have been mapped to their chromosomal locations and can be used as markers to find genes responsible for disease. The development of the genetic map (chromosomal markers), followed by the physical map, has tremendously accelerated the efforts of investigators to identify genes responsible for disease.
DNA Variation Responsible for the Diverse Human Phenotypes
With the availability of the human genome and other animal and plant genomes, we are beginning to get a glimpse of the similarities and differences between man and animal species such as the chimpanzee. In fact, the difference in the DNA sequence between chimpanzees11 is only 3% and that of the mouse12 only 4%. Another important goal that has emerged is to determine the sequences that determine phenotypic differences among human beings. Perhaps one of the surprising findings is that 99.5% of the DNA sequence is identical across all human individuals.13 This would indicate all differences in physiological attributes such as eye color, skin color, and susceptibility to disease are determined by only 15 million of the 3 billion bp of the human genome. The DNA sequences responsible for these differences include single nucleotide polymorphisms (occurring in >1% of the population), rare mutations (<0.1% of the population), short repeats of 2 to 5 bps (usually not related to disease), and large structural variants of hundreds of thousands of bps. The variation in human DNA sequences thought to be associated with common diseases are represented primarily by two categories referred to as copy number variation (CNV) and single nucleotide polymorphisms (SNP). Further evidence strongly suggests that only 20% of variation is due to CNV with the remainder due to SNPs. Our knowledge of both categories and their relationship to physiological attributes remains incomplete but has expanded rapidly in the past 2 years.14 The mapping of the location of the sequence variations determining their function and how they affect disease is a major goal for the future.
The CNVs are often referred to as structural variants because they include large chunks of DNA. Redon15 defined CNV to include DNA that is at least 1000 nucleotides but may be as large as 500,000 nucleotides. The changes may involve deletions, insertions, substitutions, rearrangements, or duplications. Since CNV is contained within large stretches of DNA, it can involve a significant proportion of the genome. Genome-wide maps of CNVs available in the public domain by Wong et al16 and by Redon et al15 contain more than 20,000 human CNV from approximately 6200 chromosome regions involving 18% of the genome.17 Several CNVs are known to induce disease and are particularly prevalent as a cause of congenital heart disease. In a recent finding, a CNV was shown to be responsible for tetralogy of Fallot.18 More than 1000 genes have been identified to be in the regions identified as CNVs. The current GWAS studies utilize a micro array that includes one million probes for CNV along with the one million SNPs. Nevertheless, it is estimated that only about 17% of common diseases are due to CNV polymorphisms.14
Following the completion of the sequencing of the human genome, several sequencing projects were pursued including the genome of the mouse,12 rat,19 and chimpanzee.11 These genomes have provided a rich resource from which to define the function of genes20 and is now part of a highly specialized science referred to as bioinformatics21 (discussed in the Bioinformatics section).
A notable project that is particularly germane to finding genes that affect health and disease is the International HapMap Consortium.22 Funded and performed by five countries, the study was initiated in 2002 and phase I was completed in 2005, phase II in 2006, and phase III in 2007. It is recognized from the sequencing of the human genome and subsequent studies that the DNA sequence is 99.5% identical.
The investigators of the HapMap Project23 for their genome-wide scans selected 1,007,329 SNPs spanning the genome. The SNPs were selected with a minor allele frequency (MAF) of ≥0.05 and for convenience was referred to as common SNP. They selected five ethnic groups for a total of 269 individuals as listed in Table 10–2. Of the 1,007,329 SNPs, 11,500 were encoding regions that specifically code for different amino acids. In addition, for comparison, 10 representative regions of the human genome of 500 kb each (totaling 5000 kb) were selected from the ENCODE (Encyclopedia of DNA Elements) Project24 and sequenced in 48 individuals. All SNPs known and unknown, common and uncommon, were sequenced and subsequently genotyped in the complete set of 269 DNA samples.
The results were very insightful and are likely to remain a landmark for decades to come. They showed the extent of LD for SNPs was greater at the centromere and less at the telomere and overall correlated with the length of the chromosome. There were several hot spots where recombination occurs. It was estimated that 80% of recombination occurs in 15% of the sequence. Hot spots typically span approximately 2 kb and are rich in the sequence motif of CCTCCCT as well as the THE1A/B retrotransposon-like element. Most of the human genome is contained in blocks. It is estimated that over one-half of the human genome is in blocks of 22k in Africans and 44k in Americans, Europeans, and Asians. In the haplotype map of the International Consortium, it was observed that the average length of the DNA block in a European/American population was 16.3 kb, in the African population 7.3 kb, and in the Chinese population, 13.2 kb as determined by one method; and by the four-gamete method, it was 5.9 kb, 4.8 kb, and 5.9 kb, respectively in these populations. Again, it was confirmed that the average number of commonly occurring SNPs (MAF ≥.0.5) in these blocks was 4 to 5. Nevertheless, there is marked variation in the size of the blocks from 1 kb to 97 kb. Within each block, it requires on average 3 to 5 common haplotypes to capture 90% of the sequence. Almost all common SNPs showed a good correlation with one or more rare SNPs. If one selected sets of SNPs, it might be possible to detect more than 90% of genetic variation. The HapMap Project classified the SNP arbitrarily into rare SNPs (MAF <5%) and common (MAF ≥5%). The GWAS described in Chap. 81 are designed to detect common SNPs and more than 100 have been identified to be associated with disease. However, they account for much less of heritability than expected. This could mean there are many more common SNPs to be identified or the rare SNPs which are expected to be associated greater effect may provide most of the heritability. The rare SNPs will require genome-wide DNA sequencing which is still financially and practically difficult. It must be noted that although each individual has approximately 3 million SNPs, they are selected by your parents from a population pool of over 17 million SNPs. The update from the HapMap recently annotated over 3 million SNPs.
The next iteration of understanding human genetic variation will be determined by developing a deeper and more ethnically divergent pool of human genome sequences. This effort has now begun in earnest using enhanced sequencing technology. Older sequencing methods, referred to as Sanger sequencing, are being replaced by high throughput next generation sequencing that can produce large scale genomic data at a fraction of the cost. Using this technology, a cooperative project has begun to establish whole genome sequence for 1000 unrelated individuals. The 1000 Genome Project is expected to establish the degree to which variation is common or rare.25
Until recently, genes implied DNA sequences that translate into protein. The traditional dogma was from transcription via translation to protein. This is still the pathway responsible for the expressed phenotype. Another layer of gene regulation has been discovered hidden in noncoding RNAs (ncRNA). These ncRNAs are transcribed from intergenic sequences and through specific mechanisms regulate transcription and/or translation of the mRNA into protein. Thousands of these ncRNAs have been identified and some of their functions have been delineated. Further understanding of their function and importance is a dynamic part of current research in genetics and molecular biology.
A gene is a distinct segment (Fig. 10–1) of the DNA forming a chromosome that has the appropriate DNA sequences promoting transcription to RNA, and the coding sequences to be transcribed and subsequently translated into a single polypeptide (see Fig. 10–1). This is the definition of a protein coding gene as illustrated in Fig. 10–2. A gene is broadly defined as having three components, the 5′ end which has most of the regulatory nontranscribed sequences, the protein coding sequences or reading frame and the 3′ end which contains primarily stabilizing sequences along with some regulatory elements. The start site for the protein coding sequences, referred to as the reading frame, is always an ATG triplet. A portion of these sequences will ultimately exit the nucleus to serve as the template to which amino acids bind to form the polypeptide or protein. As illustrated in the diagram, only a small part of the initial RNA transcript exits the nucleus as mature mRNA. These sequences that exit the nucleus and forms the template for protein synthesis are referred to as exons and those remaining behind are referred to as introns. The sequences preceding the ATG start site, referred to as the 5′ end are not transcribed but contain promoter sequences to which transcription factors bind to promote and initiate transcription. These promoters are generally classified into three categories; constitutive promoter element, enhancer elements which increase transcription, and the silencer element which turns off transcription. The other end of the gene, referred to as the 3′, may contain promoter sequences but in particular, provides stability for the messenger RNA through its AATAAA-binding tail. The 3′ end is also not translated into protein. The codons TGA, TAA, or TAG are stop codons at the end of the reading frame which terminate the translation into amino acids. Each gene requires many proteins (transcription factors) to initiate and promote transcription. These transcription factors bind to the DNA sequences of the promoter, enhancer, and silencer elements.
Figure 10–1
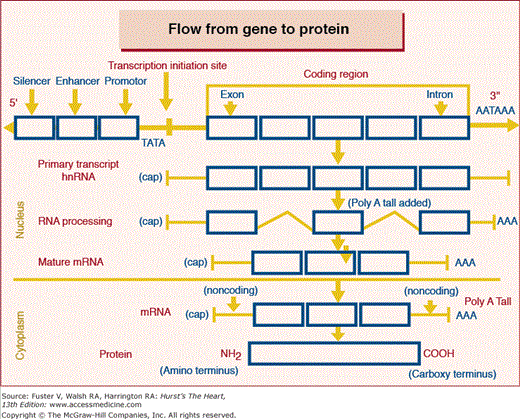
Structure of a gene and its regulated expression in the protein. The site for initiating transcription is usually indicated by the sequence TATA which is 25 to 30 base pairs upstream from the first exon to be transcribed. The RNA transcribed is then processed whereby the introns are spliced out and those remaining exit the nucleus as the mature mRNA. This serves as the template designated by each triplet of basis which amino acids are to be formed in the synthesis of the protein polypeptide.
Figure 10–2
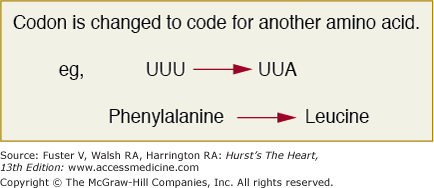
The codon UUU encodes for phenylalanine and with the mutation which changes a single base, the codon changes to UUA which happens to be a codon for leucine. This represents a point substitution mutation which alters a single amino acid and is referred to as missense. The remainder of the protein will be normal and whether this change in one amino acid alters its function will depend on whether it falls into a conserved functional domain.
Until 10 years ago, it was assumed that about 15% of the genome was involved in transcribing genes most of which translated into proteins.
A major turnaround has occurred with the discovery of DNA being transcribed into noncoding RNAs such that 93% of the genome is now believed to be transcribed and functional.26,27 A detailed review of this can be found in a recent review.28
There are several forms of noncoding RNA (ncRNA), which include short interfering RNAs (siRNAs), microRNAs (miRNAs), piwi RNAs, (piRNAs) intermediate and long noncoding RNAs (lncRNAs). All of these RNA genes are encoded in the DNA with start and stop sites, including promoter sequences similar to the genes transcribed into mRNAs. The mRNA is the intermediate step to the formation of polypeptides to form proteins. However, in ncRNA there is no messenger RNA and no protein is formed leaving the ncRNA transcript to perform its functions directly. There is extensive data to indicate that these RNAs represent a major regulatory network which through direct and indirect mechanisms control the expression of most if not all protein coding genes. Thousands of these ncRNAs exist in plants, animals, and humans. While initial studies dealt primarily with the short interfering RNAs, there is rapidly accumulating evidence for long and intermediate ncRNAs. These RNAs have been shown to act at all regulatory levels including pre-transcription, transcription, posttranscription, pretranslation, and posttranslation. These ncRNAs, through mechanisms as yet undetermined, recruit histone-modifying complexes and DNA methyltransferases to regulate and modify gene transcription. The lncRNAs have been shown to play a major role in epigenetic control of organ development.29 It has been shown that 231 lncRNAs associated with human HOX gene clusters are expressed during development.30 The small ncRNAs have been consistently linked with heterochromatin formation via the siRNA, piRNA and miRNA pathways.31,32,33 Long ncRNAs have been shown to contribute to T-cell receptor recombination and X-chromosome inactivation.34,35 Noncoding RNAs regulate transcription by interacting with transcription factors or DNA itself. miRNAs36 have been shown to induce36,37 or repress transcription.38,39 The evidence for ncRNA in silencing translation is most abundant. There are numerous examples of siRNA and miRNAs regulating translation by targeting and degrading the mRNAs. Recent studies in the mouse model of myocardial infarction40 indicate that miRNA-92 is a major controlling factor in angiogenesis and recovery from myocardial ischemia. Similar studies have been performed showing the role of miRNA in cardiac hypertrophy.41 The recent mapping of the 9p21 risk factor for coronary artery disease42
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


