Genetic Aspects of Congenital Heart Defects
Elizabeth Goldmuntz
Melissa L. Crenshaw
Since the original publication of this chapter, the medical and surgical management of children born with congenital heart defects (CHDs) has continued to progress such that most patients with significant heart disease now survive into adulthood. Simultaneously genetic technologies have advanced with remarkable speed providing an unprecedented amount of data that still begs for interpretation and understanding but has markedly increased the opportunities for diagnosis, management, and research. With these advances come new questions about variability in clinical outcome, long-term survival, fetal intervention, and recurrence risk. Although differences in clinical management affect clinical outcome and survival, evidence suggests that the etiologic basis of these malformations contributes to outcome as well (1,2,3,4,5,6,7). These observations highlight the importance of understanding the causes of CHDs so that novel interventions and preventative measures can be developed.
Environmental, genetic, and stochastic factors contribute to the cause of CHDs (Table 3.1) (8,9); an expanded discussion of environmental risk factors and teratogens can be found in Chapter 2. Evidence for a genetic contribution comes from several observations. First, specific types of CHDs are commonly seen in the context of specific chromosomal abnormalities. For example, atrioventricular septal defects are most commonly diagnosed in patients with trisomy 21, and patients with trisomy 21 are commonly diagnosed with atrioventricular septal defects. Second, similar CHDs can occur in multiple members of a family, suggesting a genetic basis. Recent studies have demonstrated particularly high heritability (implying a high genetic contribution to the cause of a disease) in certain subsets of CHDs such as left-sided lesions (10,11,12,13). Third, epidemiologic studies demonstrate an increased precurrence and recurrence risk for CHDs in families with one affected member (14,15,16,17).
Given the historically low reproductive fitness associated with severe types of CHD, it is likely and indeed evidence suggests that a portion of CHD is caused by de novo deleterious genetic mutations that do not persist in the general population (18). However, evidence also suggests that CHDs result from complex genetic and environmental interactions rather than single gene mutations or simple Mendelian inheritance. For example, although the vast majority of patients with Down syndrome have a complete third copy of chromosome 21, only 40% to 50% of patients have CHD. In contrast to the consistent finding of intellectual disability, the less frequent finding of CHDs suggests that other genetic and/or environmental factors contribute to the risk of CHDs even in the presence of a major chromosomal alteration. Similarly, parents with one child with a nonsyndromic CHD are at increased risk of having a second affected child as compared to the general population, but the recurrence risk is significantly lower than simple Mendelian inheritance would predict.
The heterogeneous etiologic factors of CHDs make it more difficult to understand the basis of these disorders. For example, several different genetic alterations are now known to be associated with tetralogy of Fallot including trisomy 21, the 22q11.2 deletion, and JAG1 mutations to name just a few (Table 3.1; see below: Genetics of Specific CHDs). Tetralogy of Fallot is found in many other genetic syndromes and can be associated with maternal exposure to retinoic acid and maternal phenylketonuria (19). Therefore, defining the genetic alterations that contribute to the cause of specific CHDs and identifying those that affect clinical outcome is challenging.
Notable advances have begun to unravel the genetic basis of these disorders. The first edition of this text published in 1968 cited 15 genetic syndromes and conditions, including storage diseases (personal correspondence from the late Dr. George Emmanouilides, Harbor-UCLA Medical Center). More than 50 years later, this chapter lists at least 50 of the most familiar or distinctive malformation syndromes characterized in part by CHDs and associated with an identifiable cause. As discoveries are made, the number of children with defined genetic causes of their CHD will increase. The information should help the physician counsel families more accurately about recurrence risks and clinical outcome and, it is hoped, lead to novel medical therapeutics to improve clinical outcomes. Since rapid progress is likely to continue, the medical caregiver will need a firm understanding of this area to anticipate its impact on clinical medicine.
This chapter reviews the genetic basis of CHDs by providing sufficient fundamental concepts to give readers a consistent background, yet remaining a pragmatic “off-the-shelf” handbook in real clinical and educational settings. The term CHDs refers to developmental changes of the intracardiac structures and major vessels and is interchangeable with the terms cardiovascular malformation or congenital heart disease. The genetic basis of cardiomyopathies and arrhythmias, both of which may be associated with a CHD, are discussed in other chapters. Instead of listing all possible genetic syndromes associated with each CHD (which can be found in multiple on-line and textbook resources listed below), this chapter will present an alternative approach for considering the genetic causes of CHD, ordered by genetic mechanism to help the reader understand and anticipate future developments in this field. In addition, Tables 3.2 through 3.4 summarize the growing number of syndromes that are either common conditions frequently associated with any type of CHD, or uncommon syndromes associated with a distinctive pattern of CHD. The chapter concludes with suggested guidelines for the genetic evaluation of a child with a CHD.
Methods and Mechanisms
Genetic Testing
In large part, the identification of novel genetic abnormalities associated with disease has been driven by increasingly sensitive methods to detect genetic alterations. The technology to detect structural variation in the human genome (150) has progressed from microscopic to genomic analyses, and is of interest to both clinician and researcher. In the first era of chromosome analysis, the karyotype displayed the 23 pairs of human chromosomes and
detected changes in chromosome number (such as trisomy 21) or large changes in chromosome architecture such as translocations (the exchange of pieces between two chromosomes). Smaller changes such as a visible deletion or duplication of chromosomal segments were subsequently detected by new “banding” methods, such as Giemsa staining, which resulted in characteristic dark and light bands for each chromosome. More recently, fluorescence in situ hybridization (FISH), multiplex ligation-dependent probe amplification (MLPA), and microarray technologies have been used to detect smaller deletions and duplications of chromosome segments that could not otherwise be seen on a standard or even high-resolution karyotype. Identification of disease-related mutations or alterations in the genetic code for a single gene can be detected using various techniques.
detected changes in chromosome number (such as trisomy 21) or large changes in chromosome architecture such as translocations (the exchange of pieces between two chromosomes). Smaller changes such as a visible deletion or duplication of chromosomal segments were subsequently detected by new “banding” methods, such as Giemsa staining, which resulted in characteristic dark and light bands for each chromosome. More recently, fluorescence in situ hybridization (FISH), multiplex ligation-dependent probe amplification (MLPA), and microarray technologies have been used to detect smaller deletions and duplications of chromosome segments that could not otherwise be seen on a standard or even high-resolution karyotype. Identification of disease-related mutations or alterations in the genetic code for a single gene can be detected using various techniques.
TABLE 3.1 Mechanisms Contributing to Congenital Heart Defects | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Accordingly, changes in chromosome number such as trisomy 21 or Turner syndrome were among the first identified genetic causes of CHDs. Deletion syndromes such as the 22q11.2 deletion or William syndromes were subsequently recognized with the advent of new banding techniques and then FISH, MLPA, and now microarrays. The advent of microarray technology has also permitted the characterization of an increasing number of deletion and duplication syndromes such as 1p36 deletion and 8p23 deletion syndromes (Table 3.2) (151,152). Given the ability of microarrays to survey the entire genome for changes in regional chromosomal imbalances, this technology has become the test of choice in many scenarios and centers (153).
Increasingly automated mutation detection and gene sequencing techniques now identify disease-related mutations in single-gene disorders such as Holt–Oram or Alagille syndromes, and in disorders characterized by a range of phenotypes and disease genes such as the RASopathies. The advent of whole exome and whole genome sequencing has expanded the ability to diagnose children and families with predominantly syndromic cardiac conditions for which the causative gene remains unknown. The growing ability to identify disease-related mutations by gene sequencing has allowed additional clinical genetic testing in individual patients or families.
Investigative Approaches
Investigators have used a variety of approaches to identify the genetic cause of CHDs reflecting advancements made in molecular genetic technologies. Historically, the identification of a consistent chromosomal alteration on a karyotype focused where investigators looked for the genetic basis of that disease. For example, 5% to 10% of patients with Alagille syndrome were originally noted to have a chromosomal deletion involving the “p” (short) arm of chromosome 20 (see Alagille syndrome). This observation suggested that other patients might have submicroscopic alterations of that region or a mutation in a gene in that region. Further investigations found disease-causing mutations in a gene called JAG1 that mapped into the region of 20p, thereby establishing JAG1 as one of the disease genes for Alagille syndrome (154,155).
Alternatively, large kindreds with multiple affected members permit a parametric linkage analysis to map the disease gene to a chromosomal position or locus. Informative linkage analyses were responsible for identifying the disease genes in Marfan, Holt–Oram, and Noonan syndrome as well as familial cases of atrial septal defect with atrioventricular conduction blockade (NKX2.5), to name a few (see: Single Gene Disorders). The Human Genome Project has greatly simplified and accelerated the identification of disease genes once a disease locus is defined.
These aforementioned strategies to identify disease-related genes are limited by the relative rarity of large kindreds with CHDs or recurrent chromosomal changes. However, emerging chromosome microarray technologies have identified an increasing number of previously unrecognized submicroscopic chromosomal deletions and duplications, thereby defining new genetic syndromes and disease genes detailed to some extent below (Table 3.2) (151,152,153,156). Indeed, such studies have also found that CHD cases harbor an increased burden of rare copy number variants (CNVs) as compared to controls though the number of recurrent CNVs found in CHD populations remains small and the clinical application of such findings remains to be fully defined (157,158,159,160,161,162,163).
TABLE 3.2 Chromosome Abnormality Syndromes Associated with CHDs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The advent of whole exome sequencing has also shed light on the genetic architecture of sporadic CHD. Whole exome sequencing of cases with rare syndromic Mendelian disorders has helped identify specific disease-related genes at an increasing rate, such as MLL2 mutations in patients with Kabuki syndrome (97). Large-scale whole exome sequencing of sporadic cases with critical CHD identified an increased burden of functionally deleterious mutations in genes expressed at high levels in the developing heart in cases as compared to controls (18). This study suggests that approximately 10% of critical CHD cases harbor deleterious, potentially disease-related mutations, which may in part explain their disease etiology. Of further import, several of the disease-implicated genes participate in chromatin remodeling activities, implicating a new developmental pathway in disease etiology (18).
As noted, it is increasingly apparent that a large proportion of CHDs are complex in origin, resulting from a combination of genetic and environmental risk factors in any one patient or family (8,9,15). Studies to date suggest marked genetic heterogeneity in the number of disease genes associated with any one lesion, and identify a wide range of genetic alterations ranging from whole chromosome to single nucleotide changes. The extent to which common and/or rare and/or novel variants, be they single nucleotide variants, deletions or duplications, contribute to disease risk is a subject of much debate and investigation. In either case, defining disease-associated genetic variants, genes, and developmental pathways provide new opportunities for novel therapeutic and preventive strategies.
Patterns of Inheritance and Familial Risks
Assessing the risk of recurrence (the chance that an affected parent will have an affected child or that unaffected parents will have a second affected child) for a patient or their relative is an ongoing challenge (164). Historically, different study designs, variable classification schemes of CHDs, different modes of ascertainment, and evolving methods of diagnosis have made it difficult to compare studies and have complete confidence in the results. Most cases of CHD have been thought to be sporadic. Overall, low recurrence risks of 2% to 4% have been quoted for all types of CHDs with one affected sibling or parent (165,166,167,168). Studies suggest that the recurrence risk increases if more than one sibling is affected (15), although it has been unclear if an affected mother confers a greater risk than
an affected father (169,170). Recurrent CHDs within a family are often concordant, or derive from the same class of defects (168).
an affected father (169,170). Recurrent CHDs within a family are often concordant, or derive from the same class of defects (168).
TABLE 3.3 Single-Gene Disorders Associated with CHDs | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 3.4 Conditions with Presumed but Unknown Genetic Cause, or Genetic Heterogeneity Associated with Congenital Heart Defects | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Data from these studies are valuable, but must be used with some caution. Earlier studies did not consider the more recently described Mendelian syndromes and chromosomal causes of CHDs, factors that affect the overall recurrence frequency. In addition, familial cases of almost every type of CHD have been observed as has every pattern of inheritance including autosomal dominant, autosomal recessive, X-linked, or complex non-Mendelian patterns of inheritance. The observed pattern of inheritance greatly influences the risk of recurrence for any one family and must be considered for counseling purposes.
As an alternative to recurrence risk analysis, investigators from the Baltimore-Washington Infant Study calculated the rate of CHD precurrence (the number of currently affected relatives at the time of birth) (14). Their studies demonstrated increased familial disease, especially for left-sided CHDs. Left-sided obstructive defects have also been the subject of several recent large family studies. Echocardiography was performed on first-degree relatives of probands to complement extensive pedigree analysis and better assess the occurrence of CHD in each family. These reports found that 8% to 19% of first-degree relatives of patients with left-sided CHDs had predominantly concordant CHDs, suggesting that left-sided CHDs commonly result from inherited genetic traits (10,11,12,13). Of note, CHDs were identified with higher frequency in first-degree relatives of probands with hypoplastic left heart syndrome (19.3%) and coarctation of the aorta (9.4%) than in first-degree relatives of probands with d-transposition of the great arteries (2.7%) (12). Most recently, several studies confirm high heritability of left-sided lesions (implying a strong genetic component to their etiology) and marked genetic heterogeneity with evidence for possible autosomal dominant inheritance in some pedigrees (10,13,171,172,173). Studies detailing the heritability of other types or classes of CHDs have not yet been performed to provide similar data.
Collectively, these findings suggest that precurrence and recurrence rates are likely to vary between specific types of CHD and within different kindred. Therefore, instead of using population-based empiric data alone, counseling for recurrence risk for an individual family requires the consideration of multiple factors including the specific type of CHD, the presence of additional affected family members, and the presence of known genetic or syndromic risk factors.
Genetic Syndromes
Genetic syndromes are defined as a consistent pattern of malformation caused by a genetic alteration. A malformation syndrome consists of multiple structural defects that are thought to be due to a single cause, even if the suspected cause has not yet been identified (80). The cause can include genetic alterations such as changes in chromosome number, translocations between chromosomes, deletions or duplications of specific chromosomal regions, or single gene defects, or can involve a teratogen (Table 3.1). The most common genetic syndromes will be described in the following sections organized by the type of associated genetic alteration. For each syndrome the genetic basis, clinical and cardiac phenotype, diagnostic testing, natural history, and population frequency are described. A more extensive table of syndromes is provided for reference (Tables 3.2–3.4). In addition to the common conditions summarized in this chapter, there is a constantly enlarging compendium of chromosome regions associated with CHDs which have evolved from cytogenetically visible aberrations (174,175) to complex genomic-based networks (176). Genetic textbooks, chapters, and on-line services provide extensive descriptions of each syndrome (80,81,177,178). An alternative listing of syndromes by cardiac subclasses may be more practical for the cardiologist searching for information based on the specific type of heart defect, especially when a dysmorphic child lacks a specific syndromic diagnosis (126).
Syndromes Associated with Chromosome Abnormalities
In population-based studies, a chromosome abnormality was detected in approximately 13% of children in the first year (27,179), and in 19% to 36% of miscarriages and stillborn fetuses with a cardiac defect (180). These occurrence data are reviewed in more detail in Chapter 2 of this text. Chromosome abnormalities can be classified according to an increase or decrease in whole chromosome number (aneuploidy), an increase or decrease of part of a chromosome (duplication or deletion, partial trisomy, or partial monosomy), or a more complex rearrangement. More recently, chromosome microarray analysis may be able to detect a causal diagnosis in up to 18% with a “syndromic CHD,” or CHD in the setting of additional congenital anomalies (181). Of the many possibilities, Table 3.2 lists the most common syndromes with the most distinctive CHDs, with selected ones discussed below.
Change in Chromosome Number (Aneuploidy)
Down Syndrome
The most familiar syndrome to cardiologists is Down syndrome in which there is trisomy 21 in 94% (a complete extra copy of chromosome 21) of cases (Table 3.2). Less commonly, (6% overall), partial trisomy of chromosome 21 is present owing to a chromosomal translocation or mosaicism. The well-known facial appearance changes with age and varies with ethnic background (Fig. 3.1). Common findings include: hypotonia, global developmental delays and moderate intellectual disability, microbrachycephaly, small
ears, mouth and nose, protruding tongue, upslanting eyes with epicanthal folds, transverse palmar creases, and sparse hair. Skeletal anomalies include fifth finger clinodactyly, brachydactyly, a gap between first and second toes, atlantoaxial instability, hypoplastic pelvis, and joint laxity. Additional problems involve the visual, auditory, endocrine, hematologic, reproductive, and gastrointestinal systems.
ears, mouth and nose, protruding tongue, upslanting eyes with epicanthal folds, transverse palmar creases, and sparse hair. Skeletal anomalies include fifth finger clinodactyly, brachydactyly, a gap between first and second toes, atlantoaxial instability, hypoplastic pelvis, and joint laxity. Additional problems involve the visual, auditory, endocrine, hematologic, reproductive, and gastrointestinal systems.
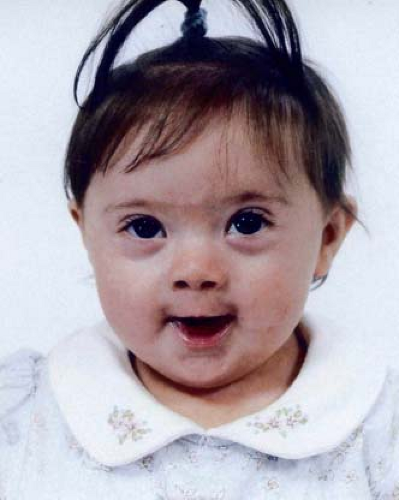 Figure 3.1 Down syndrome. A thriving 1-year-old girl with epicanthal folds, small nose, small mouth, small ears, and atrioventricular septal defect. (Courtesy of Sara S. Halbach, Donna McDonald-McGinn, Terri Anderson, and Elaine Zackai. The Children’s Hospital of Philadelphia.) |
Almost half of liveborn Down syndrome individuals have a CHD, approximately 40% of whom have a complete atrioventricular septal defect (also known as atrioventricular canal defect or endocardial cushion defect) (28). When primum-type atrial septal defect, inlet ventricular septal defect, and transitional atrioventricular septal defect are included, the frequency of the atrioventricular family of septal defects increases to almost 60% (27,28,29). The association of Down syndrome and atrioventricular septal defects is underscored by the fact that approximately 75% of patients with a complete atrioventricular septal defect have Down syndrome. Other common CHDs include secundum atrial septal defect, conoventricular and muscular ventricular septal defect, tetralogy of Fallot (with and without atrioventricular septal defect), and hemodynamically significant patent ductus arteriosus.
The risk of conceiving a child with aneuploidy (an extra chromosome), including Down syndrome increases with maternal age. Cross-sectional data (1979—2003) looking at Down syndrome children ages 0 to 19 years in 10 sections of the United States showed a steady increase from 9 to 11.8 per 10,000, but was lower among non-Hispanic blacks (182). Overall survival has improved, although prenatally diagnosed CHDs and/or growth retardation may predict a poorer outcome (183). The median age at death increased from 25 to 49 years in the interval from 1983 to 1997 (184). Equivalent if not better surgical results for atrioventricular septal defect repair with similar postoperative residual cardiovascular defects have been reported in Down as compared with non-Down syndrome individuals (185,186).
The largest survey study to date reported that the frequency of CHDs in patients with Down syndrome mosaicism was similar to the complete trisomy 21 comparison group (∼42 and 50%) (187). Atrial septal defect was the most common CHD, with atrioventricular septal defect reported in 11% compared to 22%.
The strong association of Down syndrome and atrioventricular septal defects prompted a search for a cardiac gene on chromosome 21. A Down syndrome critical region on chromosome 21 (21q22) and CHD candidate genes have been proposed, although causation for atrioventricular septal defects has not been demonstrated (188). Additional studies are searching for other genetic and environmental factors that may contribute to CHD risk in Down syndrome (189,190,191).
The only source of data on Down syndrome adults followed postoperatively (192) recommended monitoring as with any child who had a similar CHD repair. Conduction block with variable escape arrhythmia should be assessed with periodic Holter monitoring.
Trisomy 18
Although most fetuses with trisomy 18 may not survive until live birth, there are many parents who will seek aggressive support for these seriously affected children; thus, clinicians should be familiar with their appearance and CHDs (23). These children have short palpebral fissures, small mouth, micrognathia, growth retardation, prominent occiput, clenched hands, disorganized or hypoplastic palmar creases, hyperconvex nails, short sternum, small nipples, radial deficiency, and anomalies of almost every organ system.
CHDs are nearly ubiquitous and include conoventricular ventricular septal defect, tetralogy of Fallot, double-outlet right ventricle, and polyvalvar dysplasia in which two or more valve leaflets are thickened, myxomatous, or dysplastic (20,24,25). A natural history study of trisomy 18 in the United Kingdom reported that the prevalence at 18 weeks of gestational age was about 1 in 4,000, which decreased to 1 in 8,000 at birth (26). Recent population-based analyses of trisomy 18 (and trisomy 13) born during 1968 to 1999 reaffirmed that the vast majority (91%) die in the first year of life, although CHDs did not seem to affect survival (21). Although the high lethality and obligatory severe mental retardation among survivors is well recognized, some parents of trisomy 18 infants advocate for cardiac surgery, among other procedures.
There is no single trisomy 18 critical region. Instead, analysis of individuals with duplication of distal 18q provides insights into chromosome regions that may contribute to the trisomy 18 phenotype (193).
Turner Syndrome
The liveborn prevalence of Turner syndrome is approximately 1 per 2,000 (194). The phenotype depends on whether the X chromosome is absent (45,X in almost 50% of patients) or structurally abnormal (195). The most common presentation is a spontaneously aborted fetus with hydrops or lymphatic malformation in the neck or mediastinum. Fetal lymphedema produces neck webbing, protruding ears, low hairline, puffy hands and feet, and deep-set nails (Fig. 3.2). Frequent findings include short fourth metacarpals, cubitus valgus, Madelung deformity, osteoporosis, kyphoscoliosis, broad chest with apparently widely spaced nipples, renal anomalies (horseshoe kidney), nevi, hearing loss, infertility, autoimmune diseases, as well as deficits in visual–spatial/perceptual abilities, attention, and social skills. Turner syndrome women with 45,X generally have more malformations as compared to those with only partial deletion of the X chromosome. Mosaicism involving 45,X/46,XX is usually associated with a milder phenotype, and 45,X/46,XY mosaicism increases the risk for gonadoblastoma (195).
Approximately 30% of Turner syndrome women have a CHD, which usually involves the left-sided cardiac structures (30,31,195), and up to 50% of adults have vascular anomalies detected by MRI (32,196). An asymptomatic bicuspid aortic valve (15%) may progress to aortic stenosis (10%), and coarctation of the aorta (with or without a bicuspid aortic valve) is present in
10% of Turner syndrome patients. Less common left-sided defects include elongation of the transverse arch and/or pseudocoarctation (almost half of adults with Turner syndrome) (32), various mitral valve anomalies (<5%), and hypoplastic left heart syndrome (rare). Because these left-sided cardiac findings are significantly associated with the presence of neck webbing, investigators have hypothesized that the altered lymphatic drainage itself causes the associated left-sided obstructive lesions (31,197). It is unproven whether haploinsufficiency for genes on the X chromosome that may impair lymphatic and vascular development represent the underlying cause instead (33). Secundum atrial septal defect, conoventricular ventricular septal defect, partial anomalous pulmonary venous connection often involving the upper left pulmonary vein, and persistent left superior vena cava also occur, but complex CHDs, especially conotruncal defects, are notably rare (30,31,32).
10% of Turner syndrome patients. Less common left-sided defects include elongation of the transverse arch and/or pseudocoarctation (almost half of adults with Turner syndrome) (32), various mitral valve anomalies (<5%), and hypoplastic left heart syndrome (rare). Because these left-sided cardiac findings are significantly associated with the presence of neck webbing, investigators have hypothesized that the altered lymphatic drainage itself causes the associated left-sided obstructive lesions (31,197). It is unproven whether haploinsufficiency for genes on the X chromosome that may impair lymphatic and vascular development represent the underlying cause instead (33). Secundum atrial septal defect, conoventricular ventricular septal defect, partial anomalous pulmonary venous connection often involving the upper left pulmonary vein, and persistent left superior vena cava also occur, but complex CHDs, especially conotruncal defects, are notably rare (30,31,32).
 Figure 3.2 Turner syndrome. Eleven-year-old girl with hypertelorism, facial nevi, and dysplastic right pinnae. She has short stature, normal intelligence, thyroiditis, and no CHDs (Courtesy of Angela E. Lin.) |
Turner syndrome women are at risk for aortic dilation, dissection, and sudden death. Prospective MRI of older patients doubles the echocardiographic detection of aortic dilation (33% vs. 16%) (196). Arterial dilation and wall abnormalities, and cerebral involvement suggest that there may be a more diffuse vasculopathy (198,199). An epidemiologic description of aortic dissection calculated a six-fold population-based risk (36 per 100,000 Turner syndrome years), or an approximate 1.4% risk (34). Aortic dissection in Turner syndrome is almost always associated with a risk factor such as bicuspid aortic valve, coarctation of the aorta, or hypertension; the few individuals without an underlying cause may reflect inadequate examination (200,201), although an intrinsic predisposition cannot be excluded. Consensus guidelines (33) for the increasing number of older women with Turner syndrome include baseline imaging of the aorta at the time the condition is diagnosed and ongoing blood pressure monitoring. MRI may provide superior images to echocardiography depending on age, coexisting CHDs, previous surgery, and body habitus (32,196). Repeat imaging should be done every 5 to 10 years, with the appearance of hypertension, or if pregnancy is contemplated (33,202). In general, growth hormone does not appear to promote aortic dilation (35,36). A disturbing number of deaths owing to aortic dissection raises concern about the safety of pregnancy (202,203). For the increasing number of women who consider pregnancy using assisted reproductive technology with oocyte donation, it seems prudent to exclude the women who have risk factors for dissection, and plan pregnancy according to guidelines published in 2007 (33).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


