Genetic and Molecular Changes in Lung Cancer: Prospects for a Personalized Pharmacological Approach to Treatment
INTRODUCTION
Lung cancer is the most common cause of cancer-related death in men and women worldwide, responsible for over 1 million deaths annually.1 Each year, more people die of lung cancer than of the next three leading causes of cancer death combined: breast, colon, and prostate cancer. Despite advances in surgical techniques and combined therapies, lung cancer remains a disease with a dismal prognosis. Although 1-year survival has improved over the past few decades, overall 5-year survival has remained relatively unchanged at 12% to 16% over the past 30 years.2 These data underscore the need to develop new diagnostic modalities and therapeutic approaches to target lung cancer.
Lung cancer therapy is in the midst of a revolution toward personalized therapy. A key discovery in the past decade has been that some lung cancers harbor specific mutations that are essential for malignant growth (i.e., “driver mutations”), which lead to gain of function of oncogenes or loss of function of tumor suppressor genes (TSGs). In contrast, lung cancers also harbor mutations that are functionally insignificant (i.e., “passenger mutations”). While clearly promoting the oncogenic state, driver mutations are also commonly associated with “oncogene addiction,” or dependency of some cancers on one gene for the maintenance of the malignant phenotype. These dependencies, which are specific to an individual’s cancer, are absent in normal cells. Inhibition of “druggable” proteins coded for by driver mutations, such as the BCR-ABL fusion protein with imatinib in chronic myelogenous leukemia or human epidermal growth factor receptor 2 (HER2)/Neu with trastuzumab in breast cancer, are prime examples of successful therapeutic targeting of critical signaling nodes in cancer.
Historically, nonsmall cell lung cancer (NSCLC) has been classified histologically as squamous cell carcinoma, adenocarcinoma, and large cell carcinoma, and various chemotherapeutic regimens have been used to treat different histological subtypes. But with the realization that NSCLC is a collection of diseases that are identifiable by specific molecular abnormalities, personalized therapy became a goal – and now a reality – for patients with NSCLC. Between 1980 and 2000, NSCLC driver lesions that were investigated included mutations in the Kirsten rat sarcoma viral oncogene homolog (KRAS) and protein 53 (p53) genes, loss of specific chromosomal loci, loss of heterozygosity, and DNA methylation of TSGs. In 2004, driver mutations in the epidermal growth factor receptor (EGFR) gene, a membrane-bound receptor tyrosine kinase (RTK) that regulates cell growth, were discovered in NSCLC, especially in adenocarcinomas.3–5 These EGFR driver mutations resulted in a receptor with deregulated signaling driving cell growth and the oncogenic phenotype, but also led to a cellular dependence on EGFR RTK signaling. Thus, these mutations were strongly associated with therapeutic sensitivity to tyrosine kinase inhibitor (TKI) drugs that inhibited the tyrosine kinase (TK) function of EGFR. In 2007, the existence of the echinoderm microtubule-associated protein-like 4 (EML4) translocation to the anaplastic lymphoma kinase (ALK) gene resulting in an EML4-ALK fusion gene was discovered in NSCLC.6 As with EGFR, this translocation resulted in deregulated TK signaling by ALK, again driving cell growth and the oncogenic phenotype, as well as a cellular dependence on EML4-ALK signaling. Targeting the TK domain of ALK with ALK inhibitors has been found to be highly effective in lung cancers that have this translocation.
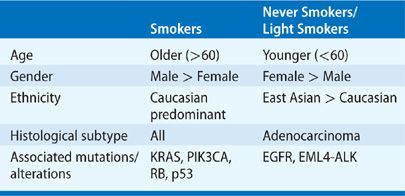
The observation that the response rates with EGFR TKIs are higher in never smokers than in smokers with advanced NSCLC led to the realization that lung cancer in never smokers is a distinct disease from tobacco-associated lung cancer, with unique molecular and biological characteristics. Tobacco components promote lung carcinogenesis through the accumulation of mutations in key genes in the growth regulatory pathways, leading to uncontrolled cellular proliferation and tumorigenesis. Although tobacco use is the oldest and the most well-established risk factor for lung cancer, lung cancer can also occur in patients who have no history of smoking. A “never smoker” is commonly defined as an individual who has smoked less than 100 cigarettes over his or her lifetime.7 The World Health Organization estimates that 25% of lung cancer worldwide occurs in never smokers,8 but in Western countries this percentage is closer to 10% to 15%.7,9–11 Histologically, the most frequent type of lung cancer in never smokers is adenocarcinoma.7 In all series of never smokers, lung cancer is much more frequent in women than in men, and generally occurs at an earlier age.7,9,12–14 Although lung cancer in never smokers occurs worldwide, geographic and ethnic variation is striking, with 30% to 40% of Asian patients with lung cancer having never smoked, compared with 10% to 20% of Caucasians.7,15–17 In the United States, African American never smokers have greater incidence and mortality rates of lung cancer compared with Caucasian never smokers.15,18 As the molecular drivers in lung cancer have been identified, it has become clear that driver mutations in never smokers are distinct from those in smokers. For example, patients with lung cancer who have never smoked are more likely to have EGFR gene mutations and EML4-ALK gene fusions, and have better response to inhibitors of these pathways than do patients with tobacco-associated lung cancer. Moreover, in patients with NSCLC, the average mutation frequency is more than 10-fold higher in smokers than in never smokers.19 Thus, it is clear that lung cancer in never smokers is a very different disease than that seen in tobacco-associated lung cancer (Table 108-1).
TABLE 108-1 Clinical and Molecular Characteristics of Lung Cancer in Smokers and Never Smokers
These discoveries illustrate that molecular biological findings can be directly linked to the development of novel chemotherapies and thereby improve the survival rates of lung cancer patients. As new pathways and mechanisms that drive lung cancer are identified, existing agents that have not been previously considered for lung cancer can now be tested, or new drugs targeting the affected pathways can be developed. These discoveries allow a patient’s therapy to be individualized to the molecular elements that drive their disease and treatment with agents that best target the characteristics of their cancer, potentially offering higher response rates and improvements in survival. Because these therapies are targeted to mutations that are only present in cancer cells, normal tissues are relatively unaffected, resulting in fewer side effects. However, hundreds of mutations occur in a cancer cell and not all of them are functionally important or suitable targets for personalized therapy. To improve the chances of success in the development of personalized medicine for lung cancer, we need to identify a functionally important target or pathway, a biomarker to identify and follow therapeutic response, the appropriate therapeutic end point, and the mechanisms of resistance to personalized therapy, both primary and acquired.
REGULATION OF CELL GROWTH AND SURVIVAL
Lung cancer results from the deregulation of cellular processes that control cell cycling and death, allowing unrestricted cell growth. Dysfunctional signaling pathways and proteins that promote carcinogenesis include membrane-bound RTKs such as EGFR, guanosine triphosphatases (GTPases) such as RAS, and nuclear proteins that contribute to cell mitosis and abrogate the appropriate apoptotic (programmed cell death) pathway such as Myc and p53, respectively.
 MEMBRANE-BOUND RECEPTOR TYROSINE KINASES
MEMBRANE-BOUND RECEPTOR TYROSINE KINASES
Membrane-bound RTKs share a common general structure composed of an extracellular region that contains a ligand-binding domain, an extracellular juxtamembrane region, a hydrophobic transmembrane domain, a cytoplasmic TK domain, and a cytoplasmic tail with tyrosine residues that serve as sites for receptor phosphorylation (Fig. 108-1). Ligand-binding results in activation of the receptor’s TK domain, leading to phosphorylation of the receptor’s tyrosine residues. This, in turn, results in recruitment of signaling molecules to the phosphorylated tyrosine residues and coupling to downstream effectors, eventually leading to biological responses. RTK activity in nonmalignant cells is normally under tight temporal and spatial regulation by the receptor and its ligand, which act as safeguards against unwanted activation. Driver lesions in the membrane-bound RTKs result in deregulated signaling or in a receptor that cannot be shut off. RTK activation in cancer may occur via overexpression of wild-type receptor due to gene amplification (resulting in excess signaling), excess ligand production, ligand overstimulation, activating mutations, translocations resulting in activated fusion proteins, or reduced receptor downregulation.
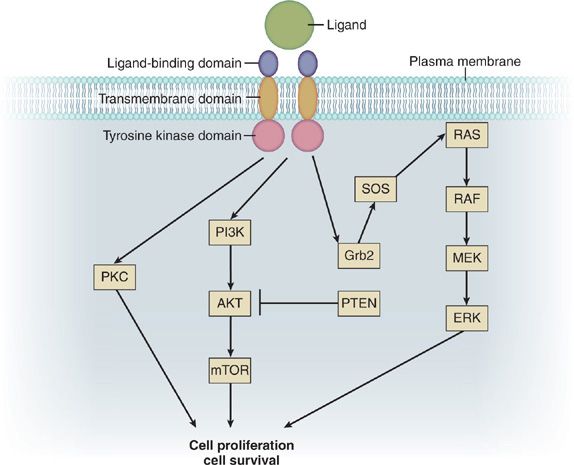
Figure 108-1 Simplified schematic of receptor tyrosine kinase (RTK) and downstream signaling pathways. Binding of ligand to the RTK leads to activation of the RAS/RAF/MEK/ERK signaling cascade, which is involved in cell proliferation. Similarly, RTK phosphorylation can also signal through the PI3K/AKT/mTOR pathway, which is involved in cell survival.
The prototypical RTKs involved in lung cancer are members of the ErbB family. The ErbB family of RTKs includes ErbB1 (EGFR; HER1), ErbB2 (HER2/Neu), ErbB3 (HER3), and ErbB4 (HER4). In the lung, these RTKs are expressed by the pulmonary epithelium (e.g., alveolar type II cells and bronchial cells) and fibroblasts. Members of the epidermal growth factor (EGF) family of peptide hormones serve as ligands for ErbB RTKs, and include EGF, transforming growth factor-α, betacellulin, and epiregulin for ErbB1; and the neuregulins for ErbB3 and ErbB4. Ligand-binding activates the receptor and multiple intracellular signaling pathways that govern fundamental cellular processes including proliferation, cell migration, metabolism, and survival. For example, phosphorylation of ErbB RTKs results in activation of growth factor receptor–bound protein 2 (Grb2) and Son of Sevenless (SOS), which ultimately leads to phosphorylation and activation of RAS. Activation of the RAS/RAF/MEK/extracellular signal–regulated kinase (ERK) pathway leads to cell proliferation. In contrast, activation of the phosphoinositide 3-kinase (PI3K)/AKT pathway by ErbB RTK signaling results in prolonged cell survival via the expression of mitochondrial antiapoptotic proteins. Thus, deregulation in an RTK pathway can give a selective growth and survival advantage to a cell, resulting in clonal expansion and tumorigenesis. Sequencing a patient’s lung cancer for EGFR or ErbB2 gene mutations can identify specific alterations in this pathway, allowing the use of targeted chemotherapy agents.
 INTRACELLULAR KINASES AND TARGETS: RAS/RAF/MEK/ERK PATHWAY
INTRACELLULAR KINASES AND TARGETS: RAS/RAF/MEK/ERK PATHWAY
Downstream of membrane-bound RTKs is the RAS/RAF/MEK/ERK pathway, which is a pivotal intracellular signaling pathway that transmits RTK signals to the nucleus via a cascade of specific protein phosphorylation (Fig. 108-1). When activated, ERK translocates to the nucleus, where it induces regulatory proteins such as the nuclear transcription factors Elk-1, Fos, Jun, activator protein 1 (AP-1), and Myc, which regulate genes that play key roles in proliferation, angiogenesis, metastasis, and chemotherapy resistance.
RAS
The RAS family of oncogenes includes neuroblastoma RAS viral oncogene homolog (NRAS), KRAS, and Harvey rat sarcoma viral oncogene homolog (HRAS). KRAS is the gene that is most commonly involved in lung cancer. RAS genes encode a family of membrane-bound GTPases, or G proteins, whose signaling regulates cell growth, differentiation, and apoptosis. Similar to RTKs, G proteins can be thought of as “on/off switches” for signaling (Fig. 108-2). In the “off” state RAS is bound to the nucleotide guanosine diphosphate (GDP), while in the “on” state it is bound to guanosine triphosphate (GTP). A phosphate group is then transferred from GTP to a RAS effector molecule, activating the downstream signaling process. The remaining GDP is incapable of signaling, and thus puts the RAS complex in an “off” state. Activation and deactivation of RAS and other small G proteins are thus controlled by cycling between the active GTP-bound and inactive GDP-bound forms. Activated RAS stimulates RAF protein kinases, which in turn initiates the MEK cascade leading to ERK activation and ultimately activation of transcription factors that regulate cell cycle progression. Other effectors of activated RAS include signal transducer and activator of transcription (STAT) and PI3K signaling cascades.
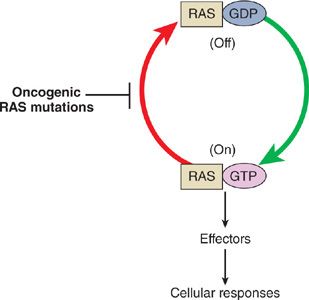
Figure 108-2 Activation cycle of Ras proteins. Growth factor stimulation leads to activation of RTKs, which causes the adaptor protein growth factor receptor–bound protein 2 (Grb2) to bind to Ras and recruit Son of Sevenless (SOS), which stimulates the conversion of Ras from the inactive guanosine diphosphate (GDP)-bound form to the active guanosine triphosphate (GTP)-bound form. Stimulation of Ras activates a mitogen-activated protein kinase cascade, leading to cell growth and differentiation. Oncogenic Ras mutations result in compromised GTPase activity, causing mutant RAS to favor its active GTP-bound state. Therefore, mutant RAS results in continuous signaling even in the absence of growth factor stimulation, conferring a proliferative advantage to tumor cells.
Approximately 20% to 30% of NSCLC harbor activating point mutations in RAS genes.20–23 Mutations occur most frequently in the KRAS gene (90%), specifically in codons 12, 13, and 61.24 These mutations result in compromised GTPase activity, causing mutant RAS protein to favor its active GTP-bound state. Therefore, mutant RAS protein results in continuous signaling even in the absence of growth factor stimulation, conferring a proliferative advantage to tumor cells.
RAF
Next in the RAS signaling cascade is RAF, which is a family of serine–threonine kinases that includes A-RAF, B-RAF, and RAF-1 (also known as C-RAF). Although RAF is activated by RAS, activation can also occur independently of RAS through other activators such as Src, c-Jun NH2-terminal kinase, interferon-β, protein kinase C-α (PKC-α), or through dimerization between RAF isoforms. Activating mutations of the B-RAF gene have been reported in up to 3% of NSCLC tumors, most of which are adenocarcinoma.25,26
ERK/MEK
To date, no activating mutations have been identified in ERK or MEK genes. However, activation of ERK/MEK signaling is a result of upstream pathway activation, and tumor cells often have coactivation of ERK/MEK and either RAS or RAF.27
 SERINE–THREONINE KINASES
SERINE–THREONINE KINASES
Membrane-bound RTKs also couple to serine–threonine kinases, which are a second class of regulatory molecules (Fig. 108-1). These enzymes also function through phosphorylation and subsequent activation of their substrate, but unlike RTKs, they phosphorylate serine and threonine residues instead of tyrosine.
Protein Kinase C
Protein kinase C (PKC) is a major component of downstream signaling pathways activated by various RTKs, including VEGF receptor (VEGFR), EGFR, and c-MET. PKC transduces signals that initiate cellular events important for carcinogenesis, including cell growth, cell cycle progression, drug efflux, apoptosis, and angiogenesis. PKC-α and PKC-β isoforms are highly expressed in approximately 20% of NSCLC tumors.28,29
PI3K/AKT/mTOR
The PI3K pathway is a complex pathway with numerous feedback loops that is fundamental for cell development, growth, and survival. Activation of the PI3K pathway is also important in the development of lung cancer. Many cell surface receptors activate second messengers that in turn activate PI3K. In addition, activation of the PI3K pathway can occur through loss of phosphatase and tensin homolog (PTEN), a lipid phosphatase that inactivates phosphatidylinositol (3,4,5)-triphosphate (PIP3). Loss of PTEN expression by immunohistochemistry or quantitative real-time polymerase chain reaction (PCR) occurs in up to 73% of NSCLC patients.30–33 PI3K generates phosphorylated phosphatidylinositides (e.g., PIP3) that bind to phosphoinositide-dependent kinase, which subsequently phosphorylates AKT at the threonine 308 residue. The resultant activation of AKT in this pathway promotes cell survival through inhibition of apoptosis and promotion of cell cycle progression, and leads to phosphorylation of mammalian target of rapamycin (mTOR), which is a serine–threonine kinase that mediates cell growth and proliferation.
By sequencing RAS, RAF, PI3K, mTOR, and PTEN genes, a specific mutation in this signaling pathway can be identified, leading to tumor-specific targets for personalized therapy. In addition, knowledge of where driver mutations occur in a signaling pathway is critical to define appropriate therapy. For example, identification of a KRAS driver mutation would suggest that therapy targeted to molecules upstream of KRAS, such as EGFR, HER2, or c-MET, would be ineffective. Clinically, this has been well confirmed.
 CELL CYCLE REGULATION
CELL CYCLE REGULATION
In nontransformed pulmonary epithelial cells, cell division is a tightly regulated process with multiple checkpoints that assess extracellular growth signals, cell size, and DNA integrity (Fig. 108-3). Cyclins and their associated cyclin-dependent kinases (CDKs) are the central machinery that control cell cycle progression. Alterations in these proteins may lead to a bypass of the checkpoints that assess a cell’s readiness to divide. These are critical checkpoints, because any mutation that is not identified and corrected by DNA repair mechanisms will be incorporated into the genome through DNA replication during S (synthesis) phase. At the next replication cycle, the cell will “read” this mutation as normal and it is subsequently fixed into the cell’s genome.
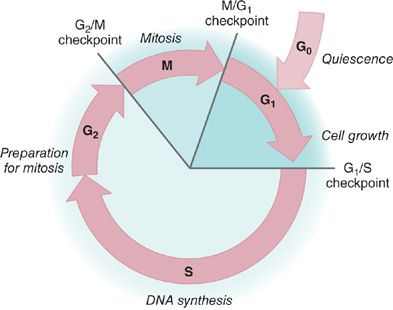
Figure 108-3 Cell cycle regulation. The cell cycle is an ordered process of events that occurs in four stages. During the two gap phases, G1 and G2, the cell is actively metabolizing but not dividing. In S (synthesis) phase, the chromosomes duplicate as a result of DNA replication. During the M (mitosis) phase, the chromosomes separate in the nucleus and the division of cytoplasm occurs. There are checkpoints in the cycle at the end of G1 and G2 that can prevent the cell from entering the S or M phases of the cycle. Cells that are not in the process of dividing are in the G0 stage.
Rb
The product of the retinoblastoma susceptibility gene, Rb, plays a central role in the G1-S transition, and Rb is considered a tumor suppressor gene. Therefore, loss of normal function of this gene removes its suppressor effect leading to cellular transformation. In its unphosphorylated state, Rb protein prevents progression from G1 to S phase by binding the key transcription factor E2F/DP-1. Once the Rb protein is phosphorylated by the cyclin D/CDK complex, E2F is released allowing transcription of a battery of genes that regulate DNA metabolism. Mutation of the Rb gene can result in a protein that does not require phosphorylation and is constitutively activated. In this case, cell cycle arrest does not occur at the G1-S transition. Alternatively, alterations in the mechanisms that phosphorylate Rb can inappropriately activate Rb, again leading to loss of the G1-S restriction point. For example, the cyclin E/CDK2 complex is an important regulator of entry into the S phase through Rb phosphorylation. Disruption of this complex regulation by increased cyclin production or loss of endogenous cell CDK inhibitors, such as the Cip/Kip family (p21, p27, p57) and the INK4 family (p16, p18, p19), can result in unregulated progression through the G1-S checkpoint.
p53
p53 is a transcription factor that can activate the transcription of numerous genes, including the CDK inhibitor p21. Under normal conditions, p53 is rapidly degraded and therefore not detectable within the cell. Mutation of the p53 gene results in a protein that fails to bind DNA effectively. Therefore, expression of the CDK inhibitor p21 gene is decreased, and p21 protein production is decreased. p21 protein is not available to stop the entry of the cell into S phase, again resulting in unregulated cell cycle progression. Mutations of the p53 gene occur in nearly 50% of NSCLC,34,35 and abnormalities of p53 protein may play an important role in the tumorigenesis of lung epithelial cells.
Disruption of the normal regulation of cell growth lies at the heart of the events leading to lung cancer. Complex networks of regulatory factors dictate whether lung cancer cells proliferate or die. Knowledge of these regulatory networks has led to identification of key pathways involved in the development of lung cancer. A detailed understanding of the molecular changes underlying lung cancer offers the prospect of specifically targeting malfunctioning pathways to achieve more rational lung cancer therapy.
PERSONALIZED THERAPY IN LUNG CANCER
 EGFR
EGFR
Small-molecule agents have been developed to inhibit EGFR kinase signaling by blocking the intracellular TK domain (Table 108-2). The first of these EGFR TKIs, gefitinib and erlotinib, began testing in clinical trials in the early 2000s. Both gefitinib and erlotinib are orally administered agents that bind at the catalytic cleft of EGFR in competition with ATP, suppressing EGFR phosphorylation and downstream signaling.36–38 Initial randomized studies in NSCLC with EGFR TKIs in combination with standard first-line platinum-based chemotherapy were all negative. However, in the TRIBUTE trial of carboplatin and paclitaxel with erlotinib or placebo, 116 patients who reported that they had never smoked had a prolongation in survival with erlotinib treatment (22.5 months) compared to placebo treatment (10.1 months, p = 0.01), suggesting treatment with erlotinib improved survival in never smokers.39
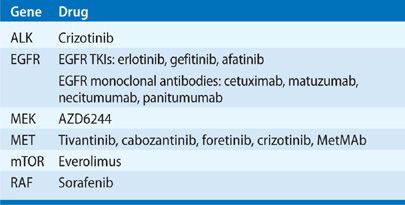
TABLE 108-2 Targeted Therapies Against Oncogenic Pathways in Lung Cancer
In 2004, as a result of basic research to understand the dramatically different response rates to EGFR TKI therapy, several academic groups reported the discovery of a collection of activating mutations in the TK domain of EGFR that correlated with a high likelihood of response to EGFR TKIs.3–5 Many of the patients harboring these mutations demonstrated rapid, nearly complete reduction of their cancers that persisted for up to several years. EGFR gene mutations were much more commonly found in female never smokers of East Asian origin with adenocarcinoma histology. Whereas the incidence of EGFR mutations in Asians is 25% to 35%, the incidence in North American and Western European patients is approximately 15%. However, the strongest predictor of EGFR mutation status appears to be absent or low smoking history.40,41
Approximately 90% of observed EGFR gene mutations are either small in-frame deletions in exon 19 clustered around the catalytic site of the receptor, or an amino acid substitution from a leucine to an arginine at position 858 in EGFR (L858R), which lies within the TK activation loop in exon 21 (Fig. 108-4).3–5,42–45 In cells harboring EGFR gene mutations, the mutated receptor leads to enhanced AKT signaling and downstream activation of antiapoptotic and prosurvival signals. The location of EGFR gene mutations within critical residues of the catalytic domain leads to altered physical structure, enhanced drug binding, and sensitivity to EGFR TKIs.46 Other types of EGFR TK domain mutations have been reported. In contrast to the more common exon 19 deletions and L858R mutation, some of these minor mutations are associated with resistance to EGFR TKIs.47–49
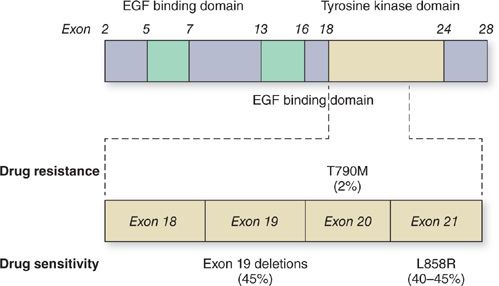
Figure 108-4 Epidermal growth factor receptor (EGFR) mutations in NSCLC. A cartoon representation of EGFR showing the distribution of exons in the epidermal growth factor (EGF) binding and tyrosine kinase domains. Exons 18 to 21 in the tyrosine kinase region where the relevant mutations are located are expanded and the most common mutations associated with drug sensitivity and drug resistance are shown.
The rapid and dramatic clinical responses to EGFR TKI therapy seen in patients with EGFR gene mutations have led to the “oncogene addiction” hypothesis,50,51 which states that because EGFR-mutant lung cancers develop in a setting in which the mutated receptor is constantly transducing high levels of antiapoptotic signals, the cancer cells are “addicted” to EGFR kinase prosurvival signaling. As a result, sudden interruption of EGFR signaling by EGFR TKIs causes massive cell death. Another hypothesis, termed the “oncogenic shock” hypothesis,52 states that proapoptotic signals are still present in EGFR-mutant lung cancer cells. When receptor signaling is inhibited by EGFR TKIs, both antiapoptotic and proapoptotic signals are blocked. However, antiapoptotic signals decay much more rapidly than proapoptotic signals, leading to a temporary predominance in proapoptotic signaling and cell death.
A large number of retrospective series have demonstrated that the response rate of EGFR mutation-positive patients to EGFR TKIs exceeds 60%,5 which is markedly higher than the typical 20% response rate to combination chemotherapy in NSCLC.53 Several studies have also shown that NSCLC patients harboring EGFR mutations survive longer with EGFR TKI therapy than wild-type EGFR patients, with median survivals of 11 to 30 months.43,54–60 Thus, personalized therapy targeting the specific genetic changes in a patient’s lung cancer is critical for improving response rates and overall survival.
Several randomized phase III trials comparing gefitinib to platinum-based chemotherapy in patients with advanced NSCLC have been reported. Although participants were not selected based on EGFR gene mutation status, the IPASS trial enriched its study population for subjects with EGFR mutations by selecting patients according to clinical factors known to be associated with higher prevalence of EGFR mutation: East Asian patients, adenocarcinomas, and never smokers or former light smokers.61 The IPASS trial was designed with progression-free survival (PFS) as the primary end point to assess the noninferiority of gefitinib compared with carboplatin/paclitaxel. The study met its primary objective demonstrating noninferiority and showed superiority of gefitinib for PFS, objective response rate (ORR), and quality of life for the entire cohort. However, results depended upon the EGFR mutation status. For patients whose tumors contained an EGFR mutation, PFS was significantly prolonged with gefitinib (9.5 months) in contrast to carboplatin/paclitaxel (6.3 months). In comparison, for those without an EGFR mutation, PFS was significantly shorter with gefitinib (1.5 vs. 6.5 months). Two additional randomized phase III trials of patients with advanced NSCLC selected for EGFR mutations (either the exon 19 deletion or L858R point mutation) also demonstrated a significant increase in PFS with gefitinib compared to platinum-based chemotherapy as first-line treatment.62,63 Similarly, a multicenter, open-label, randomized phase III trial of erlotinib versus standard chemotherapy as first-line treatment for Caucasian patients with advanced EGFR mutation-positive NSCLC (EURTAC) reported a median PFS of 9.7 months in the erlotinib group, compared with 5.2 months in the standard chemotherapy group.64 These studies confirm that treatment of EGFR mutation-positive patients with first-line EGFR TKI therapy is superior to standard chemotherapy and represents one of the most promising advances in the field of lung cancer in recent years.
Another strategy to block EGFR kinase function in patients with NSCLC focuses on the use of monoclonal antibodies directed against EGFR. Cetuximab, a chimeric anti-EGFR monoclonal antibody, has been studied in combination with different chemotherapy protocols in both phase II and phase III trials in patients with advanced NSCLC. In the phase III FLEX trial, cetuximab added to cisplatin and vinorelbine in patients with advanced EGFR mutation-positive NSCLC resulted in an absolute overall survival benefit of 1.2 months.65 Additional anti-EGFR monoclonal antibodies under study include matuzumab and panitumumab, which have been evaluated in phase II trials, and necitumumab, which is currently being evaluated in combination with chemotherapy in phase III trials.
Unfortunately, virtually all patients who initially respond to EGFR TKI therapy will develop resistance and suffer a clinical relapse. Approximately 50% of the cases of acquired TKI resistance are attributed to a secondary EGFR gene mutation, the threonine to methionine point mutation at amino acid 790 (T790M) in exon 20 (Fig. 108-4).49 The threonine residue at position 790 is highly conserved across the oncogenic RTKs EGFR, ABL, and KIT, and mutations of this residue often lead to resistance to kinase-targeted drugs.66 The T790M mutation in EGFR kinase causes drug resistance by increasing the receptor’s affinity for ATP at the ATP-binding pocket, thereby minimizing the effects of EGFR TKIs.
Several second-generation EGFR TKIs have been developed with a specific focus on the T790M mutation. Typically, these are small molecules that covalently bind to the intracellular kinase domain of the EGFR protein. In addition to binding irreversibly to wild-type EGFR, HER2, and HER4, these novel agents also bind to receptors carrying the T790M mutation. The LUX-Lung 1 trial67 studied 585 patients with lung adenocarcinoma who had progressed after treatment with a platinum-based regimen followed by at least 12 weeks of erlotinib or gefitinib, which are criteria for acquired resistance proposed by Jackman et al.68 Treatment with the second-generation TKI afatinib tripled PFS from 1.1 to 3.3 months, suggesting afatinib may have a role in the treatment of NSCLC patients with EGFR gene mutations who acquire resistance to EGFR TKIs. The LUX-Lung 3 trial investigated the efficacy and safety of afatinib compared with pemetrexed/cisplatin in patients with EGFR mutation-positive stage IIIB/IV lung adenocarcinoma.69 Treatment with afatinib led to a significantly prolonged PFS versus pemetrexed/cisplatin (median 11.1 vs. 6.9 months) and significantly higher ORR (56% vs. 23%), indicating afatinib may also be a first-line treatment option in NSCLC patients with EGFR gene mutations.
Blocking both the intracellular and extracellular domains of the EGFR kinase (“vertical blockade”) by combining a TKI and an EGFR antibody concurrently may offer an additional strategy to overcome TKI resistance. Vertical blockade of the EGFR receptor with the combination of the dual EGFR/HER2 inhibitor lapatinib and the monoclonal antibody trastuzumab has been shown to significantly improve PFS and disease control rate in HER2-positive metastatic breast cancer patients.70 In preclinical studies, dual targeting with the EGFR TKI afatinib and the anti-EGFR monoclonal antibody cetuximab was able to induce near complete responses in T790M transgenic mouse models.71 A phase 1 trial of afatinib and cetuximab in NSCLC patients with clinically defined acquired EGFR TKI resistance is currently ongoing. Further studies are underway to clarify the roles of second-generation TKIs and vertical inhibition in the treatment of EGFR TKI-resistant patients.
In addition to secondary resistances caused by T790M, several other mechanisms of resistance to EGFR TKIs have been described. MET protein overexpression or MET gene amplification was observed in up to 20% of tumor samples after treatment with EGFR TKI.72 Cells with MET gene amplification are capable of maintaining AKT activation in the presence of EGFR TKIs. In these cases, inhibition of signaling by a TKI may result in the cell switching to an alternate signal pathway, undergoing a “kinase switch” to ensure their survival. In theory, kinase switching might be counteracted by concurrent blockade of EGFR and MET kinase signaling.
 EML4-ALK
EML4-ALK
In 2007, Soda et al.6 reported a new molecular abnormality involving the fusion of ALK with EML4 in lung adenocarcinoma. The fusion protein results from an inversion in the short arm of chromosome 2 that fuses the N-terminal domain of EML4 to the intracellular kinase domain of ALK, leading to constitutive activation of the ALK kinase (Fig. 108-5). In the original report by Soda and colleagues, 6.7% of unselected NSCLC patients had the EML4-ALK fusion protein. Similar to the clinical phenotype seen in patients with EGFR mutations, the EML4-ALK fusion protein occurs more commonly in younger patients who are never or former smokers and is predominantly associated with adenocarcinoma.73,74 However, patients with EML4-ALK translocation have characteristics that distinguish them from patients with EGFR mutations. EML4-ALK-positive patients are younger than patients with EGFR mutations, with a median age of 52 years compared to 66 years.74,75 Most reports show that EML4-ALK translocations are mutually exclusive of EGFR and KRAS mutations.75–79
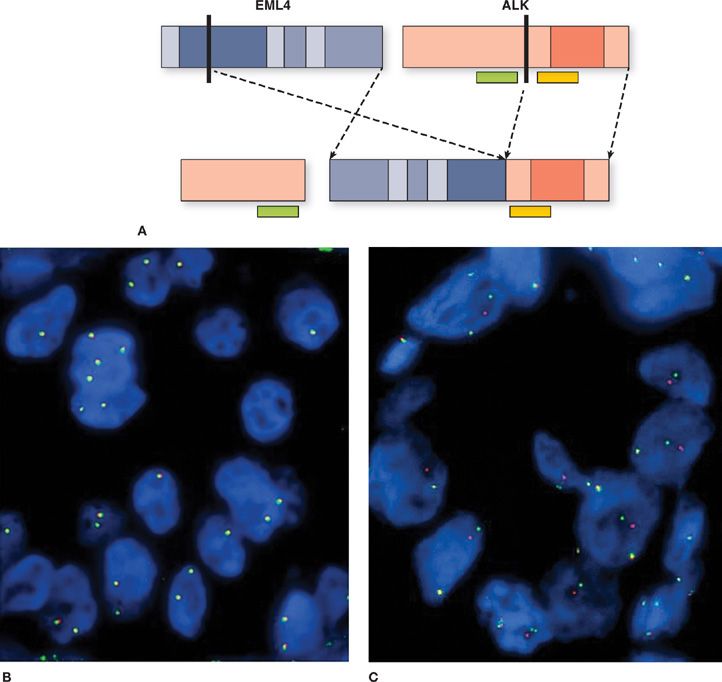
Figure 108-5 Schematic of echinoderm microtubule–associated protein-like 4 (EML4) translocation to the anaplastic lymphoma kinase (ALK) and its detection by fluorescence in situ hybridization (FISH). A. Both EML4 (depicted in blue) and ALK (depicted in red) genes are located on the short arm of chromosome 2. A chromosomal inversion leads to EML4-ALK
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


