Fig. 5.1
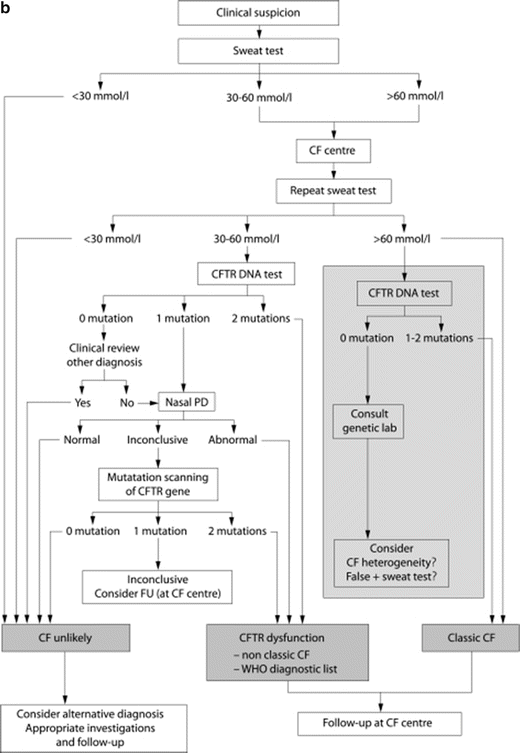
CF diagnostic algorithms. (a) North American CF diagnostic algorithm emphasizes clinical suspicion and the importance of newborn screening in the diagnostic process. Reprinted with permission from Farrell, P.M., et al., Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr, 2008. 153(2): p. S4–S14. (b). European Cystic Fibrosis Foundation Guidelines Algorithm for the diagnosis of cystic fibrosis emphasizes clinical suspicion of the disease and highlights the central role of sweat chloride testing in the diagnostic process. Reprinted with permission from De Boeck, K., et al., Cystic fibrosis: terminology and diagnostic algorithms. Thorax, 2006. 61(7): p. 627–35
A common feature in both algorithms is the central role of quantitative sweat chloride testing to confirm the diagnosis in the presence of a positive NBS or compatible clinical characteristics. Functional assays include quantitative sweat chloride as the initial test of choice, and NPD testing as an acceptable alternative in cases of inconclusive sweat chloride values or inadequate genetic analysis. Because of the central role of these CFTR functional assays, it is imperative that the basic methodology, interpretation, and limitations be understood to determine the role of these functional assays in diagnosing CF. Moreover, it is increasingly recognized that CFTR-related disorders represent a spectrum of disease, with pancreatic insufficient CF as the most severe form. It is notable that functional assessments of CFTR reflect this diversity, although the precise relationship between genotype and CFTR functional decrements can vary. CFTR biomarkers demonstrate the ability to distinguish these various phenotypes whereby increasingly severe phenotypes of clinical disease are reflected by more severe reductions in CFTR function by sweat chloride or NPD, although considerable overlap exists likely due to both biologic and environmental causes [34].
Sweat Chloride Testing
Early descriptions of derangements of CF as a salt-losing syndrome in the sweat prompted development of sweat electrolyte analysis in the 1950s [35]. Gibson and Cooke described the pilocarpine iontophoresis method in 1959 paired with a sweat pad collection system to analyze sweat electrolytes [36]. Other refinements in the test have been made since making sweat chloride analysis the most reliable method for confirming the diagnosis of CF in a positive NBS or suspected clinical syndrome [37]. Because changes in sweat chloride have been highly sensitive to use of efficacious CFTR modulators under development [38–40], sweat testing has also been frequently used as a biomarker of CFTR activity in the context of clinical trials, although data continue to emerge regarding its tissue proclivity, particularly in the context of multi-agent therapy.
Sweat Gland Physiology
The human sweat gland is composed of two regions: the secretory coil and the reabsorptive duct [37]. As isotonic secretions first produced in the secretory coil travel out the gland through the reabsorptive duct, sodium and chloride are selectively reabsorbed in the water-impermeable environment resulting in the elaboration of hypotonic sweat on the skin surface of normal subjects. In CF, the reduced function of CFTR is accompanied by downregulation of the epithelial sodium channel (ENaC), which results in diminished absorptive capacity for both sodium and chloride in the sweat gland (see Fig. 5.2) [1]. Absent or reduced function of CFTR has been linked to the chloride resorptive activity. In addition, reduced sodium transport is enhanced by diminished co-anion delivery that helps to drive low absorption. As a consequence, the sweat sodium and chloride values are elevated in CF patients compared to normal individuals. Inherent advantages of the test are its simplicity and safety, in addition to the fact that the sweat gland is not altered by CF pathophysiology, providing a reliable indicator of CFTR activity not affected by end-organ dysfunction.
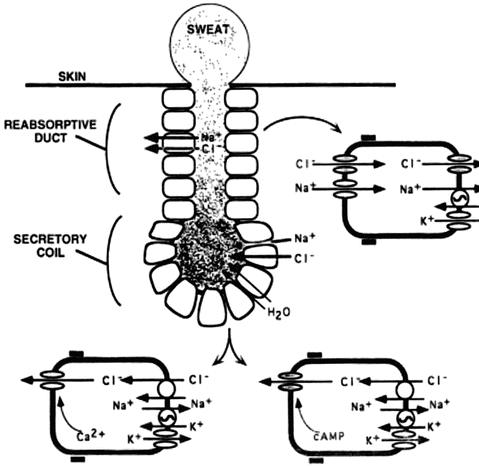
Fig. 5.2
Schematic representation of sweat production and electrolyte transport by the sweat gland. This figure shows the reabsorptive and secretory duct. The CFTR chloride channel is indicated by shading; all other channels, transporters, and pumps are indicated by open symbols. Sodium (Na+) absorption in the sweat duct occurs by sodium entry into the cell across the apical membrane driven by a favorable electrochemical gradient. Sodium then exits across the basolateral membrane in exchange for potassium (K+) on the Na+/K+-ATPase transporters, which maintains a low intracellular Na+ concentration. These processes are the same in normal and CF sweat gland ducts. Na+ transport establishes the ion concentration and voltage gradients that drive passive chloride absorption. When CFTR is defective, chloride fails to follow Na+ absorption; preventing the sodium and chloride absorption increases their concentrations in the sweat. This figure is reproduced from Welsh MJ, Ramsay BW, Accurso F, Cutting GR. Cystic Fibrosis. In: Scriver ABC, Sly WS, Valle D, editors. The Molecular and Metabolic Basis of Inherited Disease. New York: McGraw-Hill; 2001. p. 5150; with permission from The McGraw-Hill Companies
Principles Underlying the Sweat Chloride Test
An understanding of sweat gland physiology provides the basic premise underlying sweat chloride testing. Sufficient CFTR function allows for coordinated regulation of the CFTR, alternative chloride channels, and ENaC to produce hypotonic sweat. This electrolyte composition results in a low sweat chloride content. In deficient or absent CFTR function states, muscarinic stimulation of the apocrine sweat gland causes excessive chloride and other electrolytes to extrude on the skin surface because of deficient reclamation in the reabsorptive duct. Because sweat rate acts as a covariate to total ionic absorptive capacity, the extremes of sweat rate can also alter chloride values, and can be assessed independently with emerging techniques [22].
General Methods
Sweat chloride testing should be performed at accredited centers using accepted methods to avoid technical problems frequently encountered prior to wide standardization of the technique. The methods for sweat testing are described in the current standard operating procedures (available upon request from the CFF Thereapeutics Development Network) which involve three technical portions: sweat stimulation, collection, and analysis. Current methodology employs the Gibson and Cooke method of pilocarpine iontophoresis [41] which conforms to European and Australian standards [42, 43]. The preferred site for stimulation and collection is the middle portion of the extensor surface of the forearm, but successful stimulation has been achieved in other sites including the upper arm, calf, and thigh [37]. After the skin is inspected for the presence of lesions, inflammation, breaks, or abnormalities, the appropriately selected site is prepped and washed. Duplicate measurement on the contralateral forearm should be performed simultaneously when possible to ensure repeatability. After the skin is prepped, the electrodes are affixed to the skin surface with a pilocarpine-containing gel. Stimulation is achieved at a maximum of 4 mA current for 5 min, although most commercial systems are automated for stimulation. This current stimulation poses minimal risk although mild burn, urticaria, or skin irritation have been reported [44]. After stimulation, the electrodes are removed, the skin is prepped, and immediately a collection device such as the Macroduct® (Wescor, Logan, UT) is placed. Sweat is collected in this fashion for 30 min and chloride analysis conducted. In this fashion, samples can be stored at −70 °C until analyzed. An alternative method is the application of prespecified gauze to the area followed by evaluation of sweat weight and analysis of electrolyte content. Chloride concentration should be performed at a reference laboratory according to Clinical Laboratory Standards Institute guidelines. Approved methods for sweat chloride analysis include colorimetric titration (chloridimeter) and manual titration using the Schales and Schales mercuric nitrate procedure, or by automated analyzers using ion-selective electrodes [45].
Sources of Error
Successful sweat collection depends on appropriate patient selection and reducing errors in the collection process which result in the majority of errors [37]. Assuring that adequate sweat is collected and avoidance of evaporation are mandatory. Under current international guidelines which recommend use of the Macroduct® system, adequate sweat volume is achieved if ≥15 μL of sweat is collected within the 30-min collection period. The use of this system helps to minimize errors from evaporation, contamination, and loss of sweat volume, and is therefore regarded as the gold standard method of collection as it alleviates many of these sources of error. These errors are more inherent in the gauze collection method as accurate sweat rate of 1 g/m2/min is difficult to quantify by this method in order to ensure that an acceptable sweat amount is collected for analysis. In addition, because of air exposure, this method is prone to evaporative losses leading to inadequate sweat amount defined as 75 mg of sweat collected in 30 min.
Several medical conditions are associated with false-positive and -negative values for sweat chloride testing. Conditions which cause false-positive values include atopic eczema, untreated Addison’s disease, ectodermal dysplasia, glycogen storage diseases, and untreated hypothyroidism [44, 45]. False negatives from medical confounding conditions most often arise from peripheral or limb edema or, less commonly, from mineralocorticoid administration.
Because variable expression among tissues affects an important minority of CFTR mutations, sweat chloride can provide misleading information regarding diagnosis or prognosis. For example, patients with the R117H mutation can exhibit relatively preserved sweat chloride (particularly when accompanied with 7T or 9T intronic thymidine sequences), but still manifest sinopulmonary disease that is CFTR related [46]. Alternatively, splicing mutants can exhibit tissue-specific expression that impairs sweat chloride as a readout of pulmonary physiology [15, 47]. In these cases, secondary functional assessments such as NPD or intestinal current measurements have an important role to assess diagnosis and severity of functional CFTR decrements in the organ of interest.
Age at collection can affect the sweat chloride. Sweat chloride tests are falsely elevated in the first 24 h after birth, but rapidly decline thereafter [48] allowing for adequate sweat chloride collection after the first 2 weeks of infancy as a general rule. African American infants may demonstrate reduced sweat rates in the first 2 weeks of life [41, 44]. Other studies have determined that sweat values may be accurate at 48 h after birth if adequate sweat is obtained. Population studies in NBS programs from Colorado demonstrate that adequate sweat is obtained for accurate diagnosis of CF after positive newborn screen at a mean of 6 weeks after birth [49]. Advanced age may be associated with errors in the measurement of sweat chloride; a small number of studies to date have identified a trend toward increasing sweat chloride values in adolescence and adulthood that should be considered during evaluation of older individuals [50, 51]. Altered sodium metabolism can also be a confounding factor.
Diagnostic Ranges
Sweat chloride values are variable on a continuum from the lower limit of detection (10 mmol/L) to the upper limit of detection (160 mmol/L). Some controversy exists about diagnostic cutoffs as discussed in brief in the previous section, especially the “normal” cutoff value for infants, established as <30 mmol/L [52]. Generally accepted ranges for US, European, and Australian diagnostic guidelines for CF are sweat chloride values greater than 60 mmol/L and negative tests are defined as <40 mmol/L, with values from 40 to 60 mmol/L as indeterminate or borderline. These accepted intervals and interpretation are summarized in Table 5.1. These reference intervals are based on very few standardized studies to determine reference ranges [50]. Farrell and Koscik evaluated 707 subjects over a broad age range; all subjects had CFTR sequencing and clinical characteristics documented and sweat chloride testing was performed using pilocarpine iontophoresis and acceptable collection and analysis methods. A cutoff of <40 mmol/L for sweat chloride established infants as “normal,” which represented a mean plus three standard deviations. An important methodological flaw in this study was that subjects who were CFTR mutation carriers were included among normal children (n = 128), which can minimally affect sweat chloride. Other studies did not address carrier status or were performed before 1989 when the CFTR gene was sequenced [50, 53–58]. A recent infant study performed on infants 5–6 weeks of age indicates that a cutoff of ≥30 mmol/L may establish concerns for a CF diagnosis [52], and has been adopted in diagnostic algorithms for this age group. In these cases, sweat chloride is frequently monitored over the first year of life to assess whether sweat chloride abnormalities persist during development.
Table 5.1
Diagnostic ranges for sweat chloride values
Children-adult (ages >6 months) | Infants (age ≤6 months) | |
|---|---|---|
Consistent with diagnosis of CF | >60 mmol/L | >60 mmol/L |
Borderline/indeterminate | 40–60 mmol/L | 40–60 mmol/L; note that values of 31–40 have been shown to be borderline/indeterminate and require further investigation in this group (see text) |
Negative | <40 mmol/L | <40 mmol/L: see note above |
There exists a spectrum of CFTR-related illnesses from carrier status, CFTR-related disorders to CF with or without pancreatic insufficiency. The spectrum of these disorders is inadequately assessed by newborn screening or genetic analysis alone [59]. A continuum of sweat chloride values (from borderline to very high values) can be observed among various forms of disease caused by dysfunctional CFTR, from classic pancreatic insufficient CF to pancreatic sufficient CF to individuals with CFTR-related disorders and asymptomatic carriers [60, 61]. Although the ranges are broad, the differences in mean sweat chloride among these groups correlate to severity of illness and degree of CFTR dysfunction. Nevertheless, some studies demonstrate that the degree of CFTR dysfunction detected by worsening sweat chloride does not correlate to lung function severity [62], a reminder of the heterogeneity of these measurements with respect to both genetic and environmental factors and their inherent limitations.
Emerging Methods of Sweat Metabolism
Recent efforts have built upon prior assessments of sweat rate as a diagnostic tool for CF, which were originally reported in 1984 [23]. The assay takes advantage of CFTR-dependent sweating that is stimulated by β-adrenergic stimuli, as opposed to cholinergic sweating that is independent of CFTR. Evaluation of β-adrenergic sweat rate using evaporimetry has recently been introduced, and, as opposed to sweat chloride, establishes a clear decrement in obligate carriers [22]. How this test can be used to supplement CF diagnostic information will continue to emerge with additional studies.
Nasal Potential Difference
The measurement of nasal potential difference (NPD) provides a direct evaluation of sodium and chloride transport in secretory epithelial cells via assessment of transepithelial bioelectric properties [63–65]. This serves as a diagnostic aid in difficult cases where abnormal CFTR function is suspected [31, 32] or where tissue proclivity makes assessment of the airway informative. In addition, since chloride and sodium transport can be distinguished, disorders of sodium metabolism can also be assessed and reliably distinguished. NPD has been used as an important endpoint in clinical trials evaluating therapeutic agents intended to replace dysfunctional CFTR with wild-type CFTR complementary DNA (including viral and nonviral gene transfer [66–68]), restore mutant CFTR function [69–74], or address other abnormalities in CF ion transport such as novel high-affinity ENaC blockers, channel-activating protease inhibitors, or activators of alternate Cl− channels [1, 75–78].
Principles Underlying Nasal Potential Difference Measurements
The premise behind NPD measurements is that the bioelectric abnormality of the CF nasal airway reflects transport abnormalities observed in the lower airways of CF patients. In normal subjects, the bioelectric potential is maintained by a balance of sodium absorption and chloride transport, resulting in the tight regulation of the airway surface liquid volume, ionic content, and pH, which are important for maintaining mucociliary clearance and airway defense. The nasal cavity is accessible which makes it a good site to examine the ion transport characteristics of airway epithelia. Because respiratory epithelia form a tight monolayer harboring a stable and sufficient transepithelial resistance, the active secretion or absorption of charged salts such as sodium (Na+) and chloride (Cl−) ions induces a potential difference, measured as a voltage across the epithelial surface [79]. The bioelectric potential can be measured by using a high-impedance voltmeter between two electrodes, with one in continuity with each side of the epithelial surface (Fig. 5.3). The electrode on the airway surface (the exploring electrode) rests against the surface of the nasal epithelium. The internal electrode (the reference electrode) can theoretically be placed in any interstitial compartment of the body, although generally the subcutaneous tissue of the forearm is used. Subsequently, various solutions are perfused to isolate sodium transport, CFTR-dependent chloride transport, and CFTR-independent chloride transport. Due to the importance of appropriate placement within the nasal cavity and the need for an electrically quiet environment, some training and experience are required to achieve accuracy and reproducibility with the method, and are often supported by research centers within CF clinical trial networks.
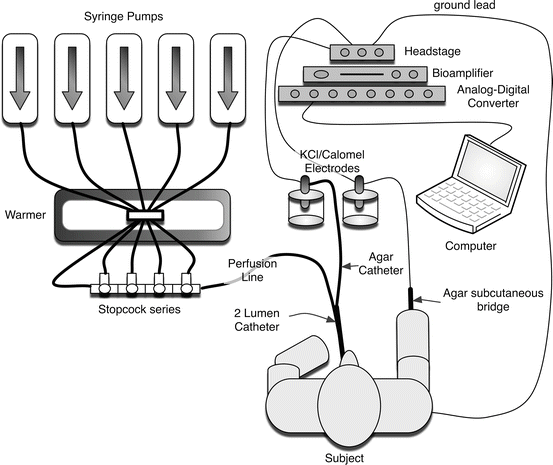
Fig. 5.3
Schematic of the nasal potential difference apparatus
General Methodology
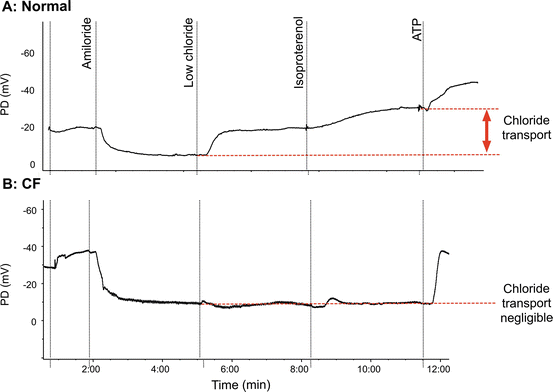
Less than a centimeter into the nose, the squamous (“skin type”) epithelium becomes ciliated pseudocolumnar epithelium under the inferior turbinate, sharing many characteristics with the more distal airways [80–82]. Under basal conditions Na+ absorption is the primary ion transport activity in normal airway epithelia [63]. The resulting (or basal) PD is negative or polarized (viewed from the epithelial surface) and in normal subjects is usually between −15 and −25 mV. The measurement continues by the sequential perfusion of compounds that block inwardly directed sodium transport through inhibition of ENaC (amiloride), followed by augmentation of chloride transport through CFTR. During perfusion of amiloride, the potential difference depolarizes as ENaC transport is reduced, and the PD typically approaches a low polarizing value (typically between 0 and −10 mV). Perfusion of a chloride-free solution induces a chloride diffusion PD through CFTR channels, resulting in a rapid and often large hyperpolarization of the PD. CFTR-mediated Cl− transport is further enhanced pharmacologically by the addition of agents known to activate CFTR, such as isoproterenol (which increases intracellular cyclic AMP as a β2 receptor agonist) [83, 84]. It is not unusual to observe transient hyperpolarization that is believed to represent Cl− transport through calcium-activated chloride channels (CaCC) during chloride-free perfusion (with or without isoproterenol). These transient polarizations typically resolve within 1–2 min, and are not CFTR dependent. Finally, ATP is perfused, which activates chloride secretion through alternative (non-CFTR) CaCCs and serves as a marker of epithelium integrity.
Healthy and CF Nasal PD Measurements
Example tracings in normal and CF subjects are shown in Fig. 5.4a, b. In CF, this ion transport profile is abnormal and the nasal PD measurement has a number of features that distinguish the PD signature. At the beginning of the measurement with buffered Ringer’s, the basal PD in CF is much more negative due to increased ENaC activity although this is also explained by increased potential in the absence of CFTR [85]. The depolarizing response to amiloride is also enhanced. Although the sodium abnormality is usually readily apparent, the most consistent abnormality in CF is the absence of hyperpolarization following perfusion with chloride-free solution and isoproterenol. In CF, the addition of ATP (or other purines) leads to a large hyperpolarization, and occurs through non-CFTR-mediated chloride secretory pathways, including stimulation of P2Y2 receptors which activate CaCCs. In aggregate, the differences in Na and Cl transport are sufficient to discriminate CF from non-CF subjects, and also detect individuals with intermediate phenotypes [34, 86].