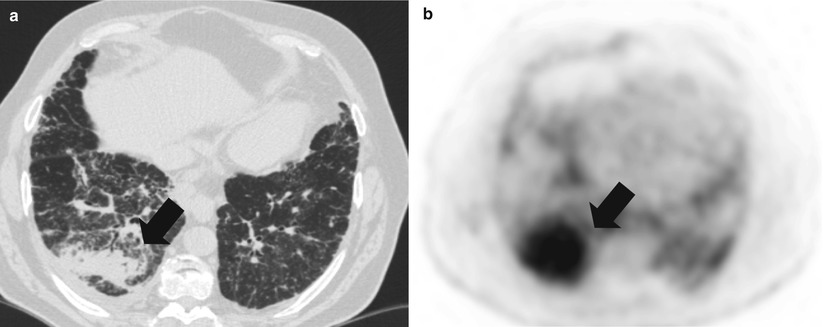
Fig. 35.1
Pneumonic-type lung adenocarcinoma. (a) Computed tomography scan of a 66 year-old former smoker woman, who presented with progressive cough. Multiple bilateral alveolar condensation-like masses with irregular margins are observed, some of which with air bronchogram. (b) 18-fluoro-desoxy-glucose positron emission tomography scan shows hypermetabolism of all the lesions. Transparietal biopsy showed adenocarcinoma cells with bronchioloalveolar and papillary architecture. No EGFR mutation was detected. The patient received platinum-based chemotherapy
Primary Pulmonary Lymphoma
Primary pulmonary lymphoma is defined as a lymphoma affecting one or both lungs, without evidence of extrapulmonary involvement or bone marrow disease at the time of diagnosis [16–18]. Primary pulmonary lymphomas are rare, representing 0.4 % of all lymphomas. Most frequent subtype is Mucosa-Associated Lymphoid Tissue (MALT) lymphoma, which is more frequently observed in patients older than 45 years, with a slight male predominance; it may also arise in younger patients with underlying immunosuppressing condition, and in patients with Gougerot-Sjögren syndrome. Less than 50 % of patients are symptomatic, with nonspecific symptoms, including cough, dyspnea, and chest pain. Unlike other lymphomas, systemic signs, such as fever, swelling, and weight loss, are uncommon. Association with immunoglobulin (Ig)M or IgG blood monoclonal gammopathy, with or without monoclonal Ig light chain secretion, possibly resulting in amyloidosis or non-amyloid monoclonal immunoglobulinic deposition disease, is observed in 30 % of cases, and may reveal the disease.
The most frequent and suggestive imaging aspect of MALT lymphoma is the “pneumonia- like” alveolar consolidation with an air bronchogram, typically localised in the middle lobe (Fig. 35.2). The “tumor-like” aspect with a solitary well-circumscribed nodular opacity is observed in 30 % of cases, with possible central air bronchogram. The “infiltrative” pattern with diffuse poorly defined ground-glass opacities, is far much rarer and may mimic, besides organising pneumonia, infiltrative lung disease and is assumed to represent early-stage disease before tumor cells invade alveolar spaces. The combination of a nodular opacity with peripheral peribronchovascular ground-glass attenuation halo is frequent. When present, associated moderate pleural effusion is suggestive of the diagnosis. FDG-PET scan may not be sensitive enough in MALT lymphoma, especially to exclude extrathoracic disease [18].
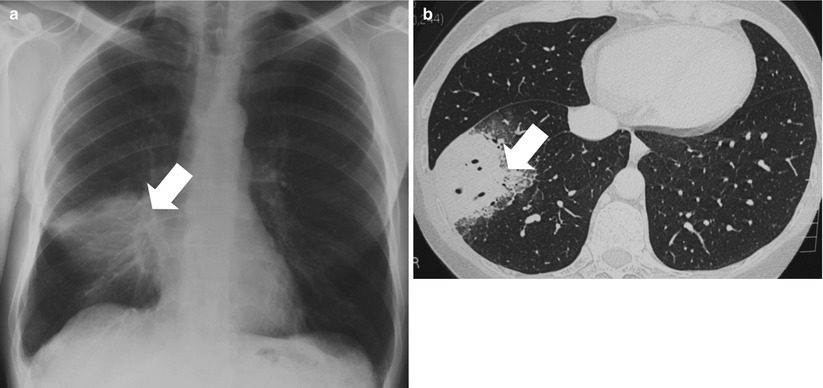
Fig. 35.2
Pulmonary primary MALT lymphoma in a 56 year-old man. (a, b) Chest radiography and computed tomography scan show persistent alveolar opacities in the right lower lobe (arrows), despite prolonged antibiotic therapy. Pathological examination of surgical biopsy showed lymphoplasmacytic-like cells of the marginal zone lymphoma associated with amyloid deposit. The patient received treatment with chlorambucil, that led to complete regression of the lesion
The diagnosis often requires a surgical lung biopsy, as cytological examination of bronchoalveolar lavage or fine-needle biopsy may show the CD20-positive B-cell infiltration but fails to exclude differential diagnoses, such as reactional lymphoid proliferation, follicular bronchiolitis, or lymphoid interstitial pneumonia [17]. Pathologically, MALT lymphoma appears as a diffuse infiltrate of small monomorphic lymphoid cells and plasmocytes, with a typical lymphangitic growth pattern spreading along the bronchovascular bundles and interlobular septa, and forming solid nodules that fill the alveolar spaces and obliterate the normal lung architecture. Immunohistochemistry is the basis of the subtype classification, with the expression of the pan-B-markers CD20 and CD79 and the absence of staining for CD5 and CD10. The proliferation is monotypic, with surface and/or cytoplasmic expression of immunoglobulin M (IgM) and, less frequently, IgG and IgA. Light chain restriction can be detected in the plasmacytic component using flow cytometry. MALT lymphomas are associated with unique chromosomal translocations, such as the (11;18)(q21,q21) resulting in a fusion of the API2 and MALT1 genes, the t(1;14) (p22;q32) involving the BCL10 and IgH genes -which is overall much less frequent, but more specific to lung locations -, and the t(14;18)(q32;q21) involving the BCL-2 and MALT1 genes [19]. These translocations may be identified in bronchioloalveolar lavage or pleural fluid specimens. In contrast with gastric and ocular annexae MALT lymphomas, no chronic infectious condition has been associated with pulmonary MALT lymphoma, although concurrent evolution with chronic hepatitis C has been described.
Therapeutic options are based on the degree of tumor extension. Surgical resection ensures both the diagnosis and the treatment of solitary nodular lesions. In asymptomatic patients, a watch-and-wait attitude may be preferred. More advanced MALT lymphoma requires more aggressive management, including single-agent chemotherapy with chlorambucil, fludarabine, and/or rituximab. Rituximab, an anti-CD20 antibody, is particularly effective and well-tolerated and may represent an alternative to chemotherapy, especially in case of t(11;18) translocation. The prognosis of MALT lymphomas is excellent, with an indolent and localised prolonged course. Local and systemic recurrences may develop in about 50 % of patients, but are usually controlled with chemotherapy. Evolution to high-grade B-cell lymphoma occurs in less than 5 % of cases.
Cancer Mimics of Interstitial Lung Diseases
Besides lymphangitis carcinomatosis, several rare intrathoracic tumors may present with an interstitial and/or a micronodular pattern, such as epithelioid hemangio-endothelioma and lymphomatoid granulomatosis.
Lymphangitic Carcinomatosis
Pulmonary lymphangitis carcinomatosis is a metastatic lung disease characterised by the diffuse infiltration and obstruction of the pulmonary parenchymal system by tumor cells [20]. Pulmonary lymphangitis carcinomatosis accounts for up to 10 % of all thoracic metastases. Patients are usually diagnosed in their fifth decade. Dyspnea is usually the chief symptom [21]. Weight loss is also frequent, as well as cough. Hypoxemia is observed in 70 % of patients.
Even if histologically confirmed, chest radiography may be normal in up to 30–40 % of cases. High-resolution computed tomography (CT)-scan may show (1) uneven thickening of bronchovascular bundles, from the hilum to periphery, that ressembles Kerley B lines, (2) a more limited or diffuse peripheral interlobular septa thickening producing polygonal arcades, and/or (3) a more aspect referred to as “beaded chain” or “string of pearls” thickening of interlobular septa [22]. These features may be diffuse or localised, uni- or bilateral, and symmetric or not. Micronodules may be observed within the thickened septa. Rapidly progressive asymmetric lymph nodes are found in 30 % of patients. 18-FDG-PET scan shows diffuse parenchymal hypermetabolism in diffuse lymphangitic carcinomatosis, or a more linear or hazy area of FDG uptake.
Diagnosis is usually obtained through bronchial, transbronchial or open lung biopsy; percutaneous biopsy may also provide lung tissue materials allowing the diagnosis to be made. Pathological examination shows multiple tumor thrombi within the lymphatic vessels, associated with a desmoplastic reaction caused by tumor proliferation and lymphatic dilatation around the interlobular septa and peribronchovascular bundles. Tumor cells of adenocarcinoma type are the most likely to produce lymphangitic granulomatosis, originating from the following primary anatomic locations: breast (33 %), stomach (29 %), lung (15 %), pancreas (4 %), and prostate (3 %) [23]. Survival is usually poor, ranging from 3 to 6 months [21–23].
Differential diagnosis includes other infiltrative lung diseases, sarcoidosis, as well as bacterial or fungal infections, such as pneumocytosis, especially in case of treatment with cytotoxic agents in patients otherwise treated for cancer [24]. Bronchoalveolar lavage is the key to make these diagnoses. Lymphangitic carcinomatosis may also be confused with drug-induced interstitial lung disease especially in patients receiving chemotherapy or biotherapy; imaging patterns may include ground-glass opacities and interlobular septa thickening. Multiple causes of interstitial lung disease may actually be interlinked in cancer patients.
Epithelioid Hemangio-Endothelioma
Epithelioid Hemangio-Endothelioma (EHE) is a low- to intermediate-grade mixed epithelioid, endothelial, and vascular tumor [25, 26]. EHE is primarily located in the liver in 65 % of patients, and the lungs are the most frequent extrahepatic location (10 % of cases); other locations include the skin and the bone [26]. EHE was initially considered an intravascular and intra-alveolar extension of bronchoalveolar carcinoma, and thus was called “intravascular bronchoalveolar tumor”. However, now it is clearly identified as a mesenchymal tumor, corresponding to low-grade angiosarcoma. Clinically, 80 % of cases of EHE occur in white females. Median age ranges from 35 to 50 years. The tumor is asymptomatic in 50 % of cases; when present, symptoms are nonspecific and include pleuritic chest pain, nonproductive cough, dyspnea, and haemoptysis [27]. Physical examination may reveal inspiratory crackles in 30 % of cases.
At imaging, EHE appears either with bilateral slow growing perivascular multiple nodules, usually located close to small vessels or bronchi, or with predominant infiltrative ground-glass opacities, with a micronodular pattern, mimicking lymphangitic carcinomatosis or infiltrative lung disease. EHE nodules usually range from 3 to 50 mm, and their number varies from 10 to 20 lesions. Nodules show increased uptake at FDG-PET scan. Nodules rarely present with cavitation or cystic features. The diagnosis may be done on either tumor lesion.
Histologically, EHE is characterised by polypoid nodules, with a central sclerotic paucicellular zone, growing into the alveolar spaces with an angiocentric distribution. Lymphangitic spread may mimic metastatic adenocarcinoma, as well as infiltrative lung diseases. Detection of the translocation t(1;3)(p36.3;q25) involving the PAX7 gene may be helpful to make the diagnosis [28]. EBV RNA sequences are detected in 90 % of cases. Overlapping entities with IgG4-related disease have been described (see below).
Although there are a few reports of spontaneous remission, the complete resection of all pulmonary nodules is the only known curative treatment of EHE. Surgery remains effective even in cases of localised recurrence. In contrast, EHE is generally insensitive to chemotherapy (cisplatin-based) or radiotherapy. Treatments with rituximab or anti-angiogenic kinase inhibitors such as sorafenib, have been reported to be effective in a few case reports [29]. In most cases, EHE is a slow-growing tumor that rarely metastasises and is associated with a median survival of 5–6 years.
The high-grade counterpart of EHE is angiosarcoma, including Kaposi’s sarcoma. No direct transformation from EHE to angiosarcoma has been reported. Clinical features are similar to EHE, but massive haemoptysis is more frequent. Radiologic features include multiple nodules with a typical surrounding halo of ground-glass attenuation, with a specific “cauliflower-like” appearance on T2-weighted magnetic resonance imaging [27]. This aspect may be shared by other disorders, including malignancies – bronchioloalveolar carcinoma, metastatic sarcomas, choriocarcinoma, melanoma, lymphoma -, infectious diseases – mycobacteriosis, aspergillosis, cytomegalovirus infection -, granulomatosis with polyangitis, and eosinophilic conditions. Management of angiosarcoma is not established: surgical resection is rarely possible owing to local and regional invasion; radiotherapy and anthracyclin-based chemotherapy is poorly effective. Modulation of immunosuppression in patients receiving immunosuppressive drugs may lead to Kaposi sarcoma remission.
Lymphomatoid Granulomatosis
Lymphomatoid granulomatosis, also called “angiocentric lymphoma,” is a malignant B-cell angiocentric and angiodestructive lymphoproliferative disorder [30]. Long considered an inflammatory granulomatous disease owing to its radio-clinical presentation that may be similar to other granulomatoses such as granulomatosis with polyangiitis (formerly Wegener’s disease) and eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome), lymphomatoid granulomatosis is now recognised as a true EBV-related lymphoid malignancy. Differential diagnosis also includes allergic bronchopulmonary aspergillosis.
The lung is the most frequent location, but the disease may also involve the brain, the skin, and the liver [30, 31]. Lymphomatoid granulomatosis occurs in middle-aged patients, from 40 to 50 years old, with a male predominance. Nearly all patients present with respiratory and systemic symptoms, consisting of cough, dyspnea, haemoptysis, chest pain, fever, and weight loss. Prolonged immunosuppression, especially in case of Human Immunodeficiency Virus infection, is a frequent underlying condition. Hypereosinophilia may be observed in the blood and/or in the bronchioalveolar lavage. The typical radiologic presentation consists of multiple smooth bilateral nodules, exhibiting a peribronchovascular pattern, ranging from 2 to 10 cm, and mainly localised in the lower lobes, mimicking multiple metastases [30, 31]. Ground-glass infiltrates may be present. Peripheral and mediastinal lymphadenopathy is absent. As in other granulomatoses, convergent nodules may migrate and form cavitated pseudotumoral masses. Pulmonary biopsy is required in most cases to make the diagnosis.
Pathologically, lymphomatoid granulomatosis forms multiple and confluent nodules composed of an atypical, angiocentric, and polymorphous lymphoid infiltration involving the vascular walls, from the subendothelium to the adventitial zones, with focal lumen obliteration. By immunohistochemistry, these lymphoid cells are characterised mostly as CD4+ T-lymphocytes, with scattered atypical cells of B-phenotype. Large B-cells are infected by EBV in 65 % of cases, a fact that correlates with the grade of the lesion.
Chemotherapy based on high dose-steroids with cyclophosphamide is the most frequently reported treatment of lymphomatoid granulomatosis. The additional use of rituximab may increase the efficacy of these cytotoxic agents [32]. However, the overall prognosis is poor, with a 5-year survival of 30–40 %, owing to progression to nodal diffuse aggressive lymphoma in 20–50 % of patients. A histopathologic grading system has been developed, based on the degree of cellular atypia and necrosis, to predict the risk of evolution to high-grade lymphoma and to select patients benefiting from early aggressive treatment [33].
Cancer Mimics of Multiple Cystic/Cavitary Lung Disorders
Cystic Tumors
Multiple cystic lung disease (MCLD) is defined by the presence of multiple rounded well-defined lucencies of low-attenuating area in the lung parenchyma that have a wall thickness lower than 2 mm [34]. MCLD may lead to the development of spontaneous recurrent pneumothorax. As discussed elsewhere in this book, MCLD may be caused, among various disorders, by lymphangioleiomyomatosis, Langerhans’ cell histiocytosis, Sjögren disease or Birt-Hogg-Dubé syndrome.
Metastastic cancers of extra-pulmonary origin may mimic, when metastasing to the lung, MCLD, especially soft-tissue sarcomas – angiosarcomas [35, 36], leiomyosarcomas [37], osteosarcomas, and synovial sarcoma [37]. Primary tumor location included soft tissues, bones, the scalp or the uterine endometer [38]. The occurrence of spontaneous pneumothoraces, which may be bilateral and recur in more than 40 % of cases, is more frequent in angiosarcoma, and is associated with poorer outcome [35]. Metastatic cysts may be associated with small-size nodules. In cases for which the information is available, pathological examination showed tumor cells in the wall of the cysts. Several observations were reported, that describe patients for whom metastatic sarcoma was ultimately diagnosed on lung biopsies, or even explanted lungs, after an initial diagnosis of lymphangioleiomyomatosis or pulmonary Langerhans’ cells histiocytosis [39].
Besides sarcomas, MCLD-like metastases have been reported in bronchioloalveolar carcinoma, metastatic or primary germ-cell tumors, colorectal and pancreatic cancer.
Cavitating Tumors
The radiological features of cavities overlap with those of cysts. A cavity is a gas-filled space, seen as a lucency or low-attenuation area, within a parenchymal nodule, consolidation area or mass [34]. Cavitation is a classical feature of pulmonary involvement by granulomatosis with polyangiitis, in combination with multiple bilateral pulmonary nodules. Cavitation is also a common feature of bronchogenic carcinoma, especially of squamous cell histology; the tumor is then usually unique in the lung parenchyma, and is not associated with extra-pulmonary manifestations in the head and neck area, the skin, the joints or the kidney [40]. Serum proteinase 3-specific, cytoplasmic antineutrophil cytoplasmic antibodies (C-ANCA) are not elevated in patients with cancer. Granulomatosis with polyangiitis rarely present with a solitary lung lesion, but most solitary necrotising granulomas are actually of infectious nature and one must have classical histology in addition to typical clinical findings to support the diagnosis.
Cancer Mimics of Pulmonary Hypertension: Pulmonary Artery Sarcoma
Pulmonary hypertension may be idiopathic or secondary to various causes, including congenital heart failure, pulmonary embolism, or collagen vascular lung diseases [41]. Historically, pulmonary hypertension was thought to result from chronic and sustained pulmonary vasoconstriction, causing shear stress on the walls of the pulmonary arteries and subsequent vascular remodeling. Many forms of human pulmonary hypertension are actually characterised by prominent muscularisation of the pulmonary arteries with partial or complete obliteration of arteries lumen by a conglomerate of cells, referred to as plexiform vascular lesion, that includes endothelial cells, smooth muscle cells, lymphocytes and mast cells [42]. Pulmonary hypertension may be observed in patients with cancer, especially in case of thrombotic or neoplastic embolism.
Pulmonary artery sarcoma presents as an endoluminal polypoid or nodular mass, which spreads along the intima of the pulmonary artery. Histologic features consist of an undifferentiated spindle cell proliferation, with marked cellular pleomorphism and high mitotic index. Leiomyosarcoma is the most frequent subtype (60 % of cases). Endovascular biopsy is feasible to obtain the diagnosis.
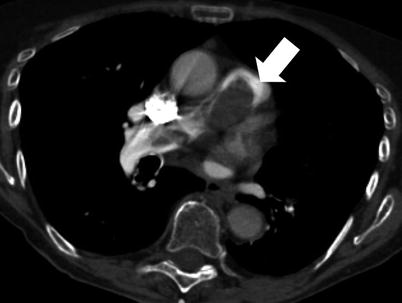
Fig. 35.3
Primary pulmonary artery sarcoma. Computed tomography scan of a 79 years-old woman, which shows complete obstruction and enlargement of the pulmonary artery trunk. Hypermetabolism is detected at 18-fluoro-desoxy-glucose positron emission tomography scan. The patient underwent pulmonary endarteriectomy, complete resection of the tumor, and extensive left pneumonectomy. The patient died 25 months after surgery
Pulmonary artery sarcomas mainly develop in patients in their fifth to sixth decade [43, 44]. Symptoms related to vascular obstruction typically mimic pulmonary embolism, with dyspnea, chest pain, cough, and haemoptysis. Failure of anticoagulants in this setting, persistence of pulmonary hypertension, as well as association to weight loss and fever (arising in 40 % of cases), may also suggest the diagnosis.
Imaging findings also help for the differential diagnosis with chronic pulmonary embolism: CT scan also shows a polypoid filling defect in the pulmonary artery, but contrary to thromboembolic disease, sarcoma forms a contiguously soft, smooth, tapering tissue, with possible extravascular nodular spread in the parenchyma (40 % of cases), and localised ground glass opacities (Fig. 35.3) [44]. Sarcoma also presents with a heterogeneous appearance including areas of necrosis and haemorrhage, and with intense hyperactivity on FDG-PET scan [45]. Magnetic resonance imaging shows T1-weighted heterogeneous enhancement, T2-weighted peripheral enhancement, and increased uptake of gadolinium, which are not found in emboli.
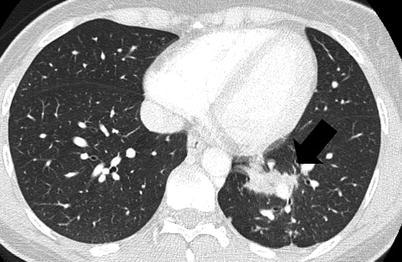
Fig. 35.4
Inflammatory myofibroblastic tumor. Computed tomography scan of a 31 year-old man who presented with persistent cough and haemoptysis following infectious pneumonia. A spiculated mass is located in the left lower lobe. Transparietal biopsy showed polymorphic inflammation without tumor cells. 18-fluoro-desoxy-glucose positron emission tomography showed focal hypermetabolism of the mass. Surgical resection was performed. The patient did not receive adjuvant treatment. No recurrence was observed after a 1-year follow-up
Surgery is the only potentially curative treatment and, even if performed in an emergency setting in case of acute right heart failure, allows resectability in 60–75 % of cases [46]. Adjuvant chemotherapy may be administered, especially in incomplete resection, with limited evidence from the literature. Contrary to soft tissue sarcomas, prognosis is mainly related to tumor location, because half of the patients die as a result of the progressive obstruction of the pulmonary trunk [45, 46]. Reoperation is feasible in 30 % of cases. However, in recent series, overall median survival is as low as 6–12 months.
Intrathoracic Pseudotumors
Pseudotumors represent a wide range of etiological, pathological, and clinical-radiological disorders, that all share some degree of reactive inflammation and may present with some cancer-related molecular hallmarks. Pseudotumors may mimic the clinical and radiological features of various intrathoracic diseases.
Inflammatory Myofibroblastic Tumor
Inflammatory myofibroblastic tumor (IMT) is the most representative entity of the pulmonary pseudotumors. The term “inflammatory myofibroblastic tumor” was proposed in 1990 by Pettinato et al. [7, 8, 47], and encompasses a wide spectrum of lesions previously called “inflammatory pseudo-tumor”, “fibroma”, “fibroxanthoma”, “fibrous histiocytoma”, “plasma cell/mast-cell/solitary granuloma,” “plasma cell histiocytoma complex” or “pseudosarcomatous tumor” [2, 7, 8, 47]. IMT is rare, with a prevalence of 0.04 % of resected pulmonary neoplasms in the surgical series of the Mayo Clinic [7]. Sub-classifying IMT meets with the fact that both benign and malignant features are observed, representing an archetype of neoplastic/non-neoplastic borderline disorder [3, 6].
Pathologic Features
IMT macroscopically appears either as an intra-parenchymatous well-circumscribed mass of variable size [47–49]. Histologically, the tumor is made of irregular proliferation of fibroblasts and myofibroblasts intermixed with an infiltrate of inflammatory cells, mainly lymphocytes and plasma cells. Three major distinct histological patterns are usually recognised [7, 8, 47–49]: (1) the “plasma cell” – also called “lymphoplasmocytary” – variant, composed of inflammatory myxoid proliferation with fascicles of spindled fibroblasts or myofibroblasts, abundant lymphocytes and plasma cells, and minimal fibrous connective tissue; (2) the “fibrohistiocytic” type, showing a compact spindle-cell pattern simulating fibrous histiocytoma, and characterised by a myxoid proliferation of fibroblasts and myofibroblasts associated with polyclonal plasma cells, xanthoma cells, and rare giant cells; and (3) the “organising pneumonia-like” type, with a hypocellular pattern characterised by dense collagen with sparse spindle cells. The proliferating myofibroblastic cells show no cellular atypia, no necrosis, and only rare mitotic figures. The myofibroblastic cells usually stain for vimentin and smooth muscle actin. Differential pathological diagnosis includes all the diseases composed of fibroblasts and myofibroblasts, some of which may overlap with IMT [8]: benign and malignant fibrous histiocytoma, myofibroblastoma, inflammatory fibrosarcoma, spindle cell carcinomas, plasmacytoma, as well as organising pneumonia.
Pathogenesis
The concept of IMT as a proliferating neoplasm has been questioned [8]. Historically, IMT – then called “inflammatory pseudotumor” – was thought to originate from organising pneumonia through exaggerated inflammatory response to injury. Older case reports emphasised that, in as many as 30 % of cases, chronic or repeated infections could be a potential cause, but this concept was reconsidered with more recent reports that included CT-scan studies, suggesting that recurrent pulmonary infections were rather a consequence of bronchial obstruction by the tumor [7, 48].
The concept of IMT as an immunologic disorder was raised after the anecdotic detection of EBV and human Herpes simplex Virus-8 sequences in myofibroblastic cells, with associated expression of cytokines such as interleukin (IL) 6, IL8 and cyclin D1 [7, 50]. The recent identification of immunoglobulin (Ig) G4 expression in polyclonal plasma cells extracted from intrathoracic IMTs led to consider that an immunopathological process may participate in the development of these tumors, especially the plasma-cell variant ones. Such proliferation of IgG4-positive cells has also been associated with auto-immune disorders including sclerosing pancreatitis, retroperitoneal and mediastinal fibrosis [51]. IMTs may then be part of IgG4-related disease, newly recognized fibroinflammatory condition characterised by tumefactive lesions, a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform fibrosis, and, often but not always, elevated serum IgG4 concentrations.
Molecularly, myofibroblasts are considered as pivotal in the development of IMT. Clonal gene rearrangements may be observed at caryotype [52], especially involving the Anaplastic Lymphoma Kinase (ALK) gene located in region 2p23 [53]. Diffuse ALK overexpression is observed in 40–70 % of IMTs at immunohistochemistry, but may also be identified -usually at lower levels- in a wide variety of non-IMT soft tissue tumors. ALK rearrangement is identified in 40–50 % of IMT cases, especially in younger patients, and most frequently consists of t(1;2)(q21;p23) translocation implicating the tropomyosine 3 gene [54]. Given the oncogenic nature of ALK activation, these data lead some authors to consider IMT as a true neoplasm. Other elements further reinforce this concept, including the presence of vascular invasion, the local recurrence rate as high as 25 % [7], the existence of multifocal lesions in 5 % of cases, reports about malignant transformation, and genomic and expression profiling data, showing DNA aneuploidy, P53 expression, and implication of other known oncogenes such as glutathione-S-transferase [55]. Overlap exists between IMT, IgG4-related disorders, and inflammatory fibrosarcoma that exhibits prominent cellular atypias and necrosis.
Clinical-Radiological Features
Clinically, pulmonary IMTs mostly occur before the fourth decade, while accounting for more than 50 % of pulmonary tumors in children [7, 48, 56]. Patients are asymptomatic in about 60 % of cases, or may present with non-specific symptoms including chronic cough, dyspnea or rarely haemoptysis. At imaging, IMT appears as a solitary well-circumscribed peripheral mass, ranging from 2 to 15 cm in size (Fig. 35.4). Contrary to extra-thoracic locations, pulmonary IMT is usually solitary. Calcifications are observed in 15 % of cases.
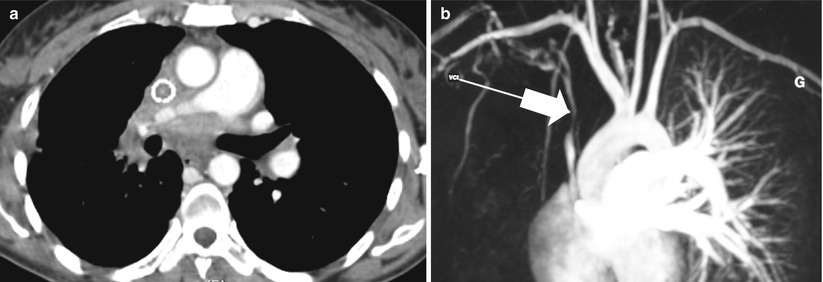
Fig. 35.5
Sclerosing mediastinitis. (a) Computed tomography scan of a 32 year-old woman who presented with progressive dyspnea and superior vena cava syndrome. Connective tissue proliferation infiltrates the entire mediastinum. Surgical biopsy was performed to make the diagnosis. (b) At magnetic resonance angiography, the calibre of the superior vena cava is reduced (arrow). An endoprothetic tube was placed as palliation
The usual stability in size over time is an important imaging feature. Multifocal and bilateral IMTs are exceptional, and are considered as overlapping forms of low-grade fibrosarcoma or malignant fibrous histiocytoma. IMTs are usually hypermetabolic at 18-fluorodeoxyglucose-positron emission tomography (FDG-PET)-imaging. Extra-pulmonary involvement occurs in 10–20 % of cases, and is mostly observed in the mediastinum and the pleura. Conversely, extra-pulmonary IMTs may metastase to the lung, especially in younger patients.
Preoperative diagnosis with endoscopic or per-cutaneous biopsies remains difficult due to the heterogeneous morphology of IMTs. Cytological fine-needle aspiration accuracy was as low as 42 % in a recent study [57].
Treatment
Even if historically considered a benign lesion with possible spontaneous regression, IMT is usually treated by surgical resection due to its tendency to grow, to provoke local complications including haemoptysis and infection, and to potentially relapse with local, pleural, parietal, or mediastinal invasiveness (15–25 % of cases and 3–5 % of cases, respectively) [7, 58, 59]. The need for adjuvant treatment in case of incomplete resection has not been evaluated. In non-operable patients, focal conformation radiotherapy or corticosteroid challenge may represent an alternative. Corticosteroids are reported to induce objective responses in as many as 50 % of cases, especially in predominantly plasma-cell tumors and IgG4-positive tumors [59]. In recurrent or multifocal lesions, chemotherapy may use the same regimens as for soft-tissue sarcomas. Crizotinib (PF-02341066, XALKORITM; Pfizer, NYC, NY), a small pharmaceutical tyrosine kinase inhibitor of ALK, was recently reported to produce tumor responses in 2 patients with ALK-rearranged extra-thoracic IMTs [60].
Prognosis
Patients with a resected inflammatory myofibroblastic tumor have a 5-year overall survival ranging from 75 to 100 % [7, 57–60]. Transformation to low- and/or high-grade fibrosarcoma has exceptionally been reported, and may correspond to initially misdiagnosed high-grade tumors [2, 58]. ALK-positive tumors are considered to be more aggressive, but local recurrence actually occurs regardless of ALK expression level [53]. The most consistent prognostic factor is actually the initial invasiveness of the tumor.
Overall, IMT, while having been considered for a long time a reactive lesion simulating a neoplasm, is now rather regarded as a true low-grade neoplasm. However, other pseudotumors are described in adults, of diverse pathogenesis [4, 6]. Moreover, several entities related to IMT have been described, including sclerosing mediastinitis and hyalinising granuloma.
Sclerosing Mediastinitis and Hyalinising Granuloma
Similar to IMT, sclerosing mediastinitis and hyalinising granuloma both consist of tissular infiltration by dense collagen fibrosis forming lamellar bands, interspersed with lymphocytes and plasma cells [61–63]. These two entities differ by the primary anatomic location: sclerosing mediastinitis predominantly involves the mediastinum, with possible extension to the lung parenchyma; hyalinising granuloma occurs within the lung parenchyma without contiguous involvement of the mediastinum. Overlap exists between these entities and other fibrosing disorders such as IMT, retroperitoneal fibrosis, and other IgG4-related disorders.
Sclerosing Mediastinitis
A hallmark of sclerosing mediastinitis is the obstruction of major mediastinal veins, with multifocal venous infarcts observed in the tumor leading to lumen obstruction by cellular fibrosis, haemorrhage, and necrosis. Sclerosing mediastinitis has mostly been described in North America, where it is thought to result in most cases from exacerbated response to Histoplasma [64]. Sclerosing mediastinitis may actually be idiopathic, familial, or associated with various disorders, including other fungal infections – aspergillosis, cryptococcal infection -, tuberculosis, or sarcoidosis. It has also been described following radiation therapy, ergot derivatives, or beta-blockers treatment. Autoimmune reaction may also participate to the development of sclerosing mediastinitis, as association with elevated serum IgG4 syndrome has been reported [65].
Clinically, sclerosing mediastinitis is observed in the fourth decade, with a slight male predominance. Patients may be asymptomatic or develop chest pain, fever, haemoptysis, dyspnea, as well as signs related to mediastinal structures invasion; venous infarcts of the lung can be the first manifestation of the disease. The amplitude of symptoms is actually related to the extension of the sclerosis. At imaging, sclerosing mediastinitis presents either as a localised and calcified mass in the paratracheal, hilar, or subcarinal areas, or as a diffuse infiltrate of the whole mediastinum, with no calcification (Fig. 35.5). The lung may be marginally affected, with consolidation areas associated with venous infarcts, and possible pleural effusion.