Extrapulmonary Syndromes Associated with Lung Tumors
A wide variety of extrapulmonary syndromes have been reported with lung cancer. This chapter will focus primarily on paraneoplastic syndromes, a heterogeneous group of disorders associated with cancer but not due to direct invasion, obstruction, or metastasis.1,2 As many paraneoplastic syndromes share similar underlying mechanisms, they may be classified as endocrine, hematologic, and neurologic syndromes. More broadly speaking, they are either hormonally or immunologically based.
Paraneoplastic syndromes affect up to 8% of patients with cancer,3 and of all malignancies, lung cancer is most commonly associated with these syndromes. The subtype of lung cancer most commonly associated with paraneoplastic syndromes is small-cell lung cancer (SCLC), but some such as hypertrophic pulmonary osteoarthropathy (HPO) and hypercalcemia are more common in non–small-cell lung cancer (NSCLC).4
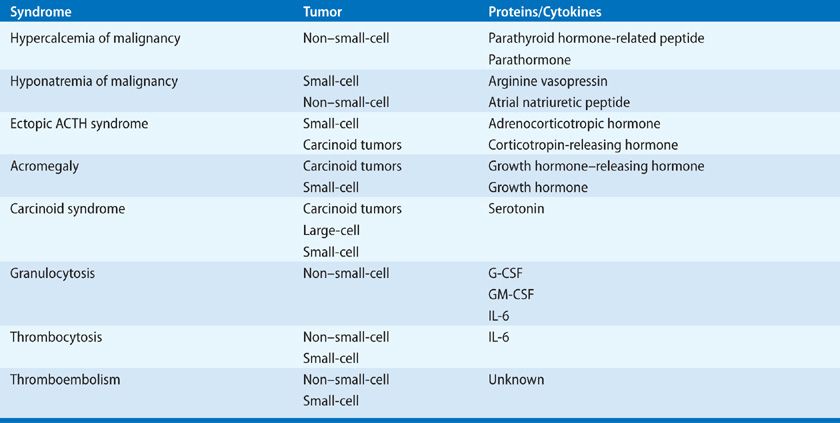
Endocrine and hematologic syndromes associated with lung tumors are listed in Table 118-1. The endocrine syndromes are characterized by the ectopic production by tumor cells of biologically active peptide hormones that bind to receptors in adjacent or distant organs, giving rise to a clinical syndrome. Hematologic syndromes develop in patients with lung cancer through the production by tumor cells of cytokines that activate progenitor cells in the bone marrow. Neurologic syndromes, such as encephalomyelitis and subacute sensory neuropathy, are caused by the induction of antibodies directed against proteins expressed by the lung cancer cells and directed against antigens present on cells in the nervous system. In clinical practice, an understanding of the extrapulmonary syndromes is important for several reasons: (1) recognition of a syndrome may serve as a harbinger of an occult malignancy or may signify disease recurrence; (2) the course of the endocrine and hematologic syndromes usually parallels the course of the lung cancer, although the neurologic syndromes frequently do not; and (3) appropriate treatment of the extrapulmonary syndrome often reduces the patient’s morbidity and may allow definitive treatment of the cancer. In general, definitive treatment of the underlying tumor is the most effective form of therapy for the paraneoplastic syndromes, so their presence should not preclude treatment of lung cancer with curative intent as most syndromes are reversible once the patient receives treatment.
HYPERCALCEMIA OF MALIGNANCY
Hypercalcemia is commonly seen in cancer patients5,6 and lung cancer is the most common solid tumor associated with it, occurring in up to 20% of patients.7,8 It is overall the most common of the paraneoplastic syndromes. It occurs most frequently in patients with squamous cell lung cancer, but is occasionally seen in patients with adenocarcinoma and very rarely in patients with SCLC.9
 BIOLOGY
BIOLOGY
Etiologic mechanisms for hypercalcemia in patients with lung cancer include parathyroid hormone–related peptide (PTHrP) production, increased levels of the active metabolite of vitamin D (calcitriol, also called 1,25-dihydroxyvitamin D3), and localized osteolytic hypercalcemia.5,10 Most cases of hypercalcemia in patients with lung cancer are caused by the ectopic production of PTHrP by tumor cells (referred to as humoral hypercalcemia of malignancy).11–13 The PTHrP gene expresses three messenger RNAs (mRNAs) that encode three distinct peptides, each differing at the COOH-terminal region (Fig. 118-1). Eight of the first 13 amino acids in PTHrP are homologous with PTH, so similar functional activity is shared between the two peptides. PTHrP mRNA and peptides have been demonstrated in cancer cells from patients with lung cancer and hypercalcemia. PTHrP has been shown to bind to PTH receptors in the bone and kidney, which causes increased osteoclastic bone resorption, decreased bone formation, and decreased calciuria, leading to hypercalcemia.14,15 Levels of 1,25-dihydroxyvitamin D3 are suppressed in patients with PTHrP-induced hypercalcemia, but are raised in patients with primary hyperparathyroidism. This difference occurs because renal α-hydroxylase activity is low in PTHrP-induced hypercalcemia, unlike primary hyperparathyroidism.16 PTH production by lung cancer cells has also been described, but it is a very rare cause of hypercalcemia of malignancy.17–19 Other factors that cause bone resorption have been identified in the plasma of patients with lung cancer, including transforming growth factor-α and a vitamin D metabolite. These are very rare, however, and their causative role in hypercalcemia has not been shown conclusively.
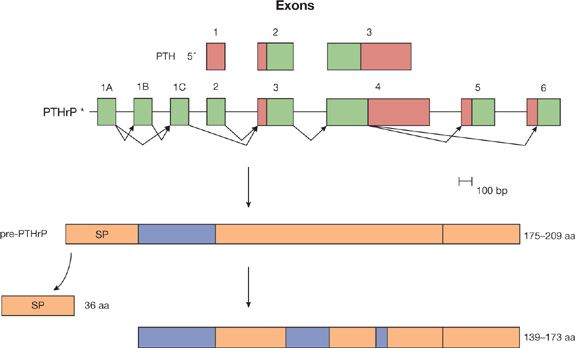
Figure 118-1 Parathyroid hormone and parathyroid hormone-related peptide. The human PTH gene has three exons, which constitute the protein-coding segments. The protein-coding segments are represented by the black boxes. The PTHrP gene is more complex, with eight exons. Through alternative splicing, three different isoforms of mRNA can be produced. These isoform mRNAs encode the pre-PTHrP proteins, which vary in size from 175 to 209 amino acids (aa). Thirty-six amino acids are removed from the amino terminal end as the signal peptide. Three different PTHrP molecules are produced, with 139 to 173 aa. The rectangular region at the carboxy terminal represents the different lengths of PTHrP. The N-terminal region (1–34 aa) mimics the classic PTH-like function (hatched box). The midregion (67–86 aa) of the peptide stimulates placental calcium transport (shaded box). The C-terminal region (107–111 aa) inhibits osteoclastic bone resorption (double hatched box).
 DIAGNOSIS
DIAGNOSIS
The early symptoms of hypercalcemia include thirst, malaise, fatigue, anorexia, polyuria, constipation, nausea, and vomiting. As hypercalcemia becomes increasingly severe (>14.0 mg/dL), confusion, lethargy, coma, and death can occur. The demonstration of a suppressed intact parathyroid hormone (iPTH) level and a low or normal calcitriol level along with an increased concentration (>10.5 mg/dL) of calcium in the serum of a patient with NSCLC should suggest this paraneoplastic syndrome. This is in contrast to the findings of elevated iPTH and calcitriol levels in primary hyperparathyroidism, which may occur in up to 10% of patients with cancer. A complete diagnostic evaluation includes measuring serum concentrations of iPTH, PTHrP,20 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D, calcium, albumin, magnesium, and phosphorus. Other potential causes of elevated serum calcium should also be excluded. Thiazide diuretics, vitamin D or lithium administration, hyperthyroidism, and sarcoidosis are potential causes.21 Bone scintiscan should be obtained to exclude bone metastases. An elevated PTHrP level in the absence of bone metastases establishes the diagnosis of humoral hypercalcemia of malignancy caused by ectopic PTHrP.15,20
 TREATMENT
TREATMENT
As with other paraneoplastic syndromes, treatment of the underlying cancer is the most effective method of treating the humoral hypercalcemia associated with lung cancer. Until this can occur, medical management of hypercalcemia must be considered. If asymptomatic or mildly symptomatic, hypercalcemic patients with calcium levels <12 mg/dL (3 mmol/L) do not require immediate treatment. Similarly, a serum calcium level of 12 to 14 mg/dL (3–3.5 mmol/L) may be well-tolerated chronically, and may not require immediate treatment. However, an acute rise to these concentrations may cause a marked change in sensorium, requiring intervention. In addition, patients with a serum calcium concentration >14 mg/dL (3.5 mmol/L) require treatment, regardless of symptoms.22,23 Treatment includes intravenous saline, with the addition of furosemide diuresis only after correction of intravascular volume depletion. Subcutaneous calcitonin has a rapid onset of action and is most useful in severe cases. Mithramycin and long-acting bisphosphonates, such as pamidronate, are effective for long-term control of hypercalcemia.24 Corticosteroids exert their effect through inhibition of dihydroxyvitamin D3 synthesis and therefore have less effect in patients with elevated PTHrP.
Humoral hypercalcemia usually develops in patients with advanced progressive cancer and is associated with a median survival of about 1 month.25,26 Nonetheless, in some cases, treatment of hypercalcemia serves an important palliative role by improving symptoms and allowing patients to be discharged from the hospital.
HYPONATREMIA OF MALIGNANCY
Hyponatremia is a frequent complication in patients with cancer. It occurs at presentation in approximately 15% of patients with SCLC and 1%of patients with NSCLC.27 The ectopic production of arginine vasopressin (AVP) by cancer cells plays a causal role in the majority of cases.27 This form of hyponatremia is recognized as the syndrome of inappropriate antidiuretic hormone (SIADH). Up to one-third of patients with lung cancer and hyponatremia do not demonstrate elevation of AVP in their tumors or plasma.28–30 In these patients, the ectopic production of atrial natriuretic peptide (ANP) has been implicated, but the exact contribution of this hormone remains to be defined.28
 BIOLOGY
BIOLOGY
AVP is a 9-amino acid peptide normally produced by the neurohypophysis. The peptide binds to receptors in the kidney to reduce the excretion of free water. When plasma osmolality exceeds 280 mOsm/kg, the release of AVP from the pituitary increases, causing the kidney to retain more free water and maintain fluid and osmolar balance. In patients with SCLC, ectopic production of AVP causes hyponatremia by inhibiting free-water excretion in the distal tubule of the kidney. AVP mRNA is expressed in SCLC cells and the peptide is translated and secreted (Fig. 118-2). Measured levels of AVP in plasma are often increased.31
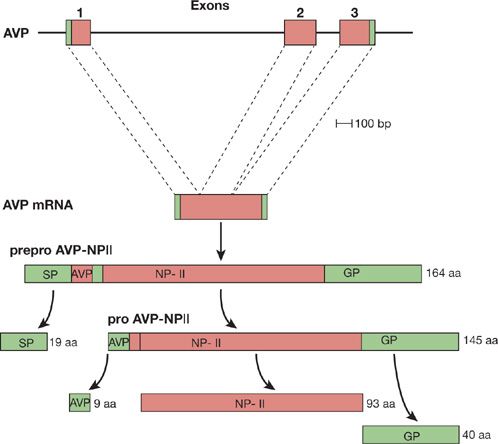
Figure 118-2 Arginine vasopressin. The three exons of the human AVP gene rise to a 700-base arginine vasopressin mRNA. The mRNA is translated into a 164-amino acid (aa) preprohormone with a 19-amino acid amino terminal signal peptide (SP). The SP is cleaved, giving rise to a 145-amino acid prohormone. This prohormone is processed into the nonapeptide AVP, a 93-amino acid neurophysin (NP), and a 40-amino acid glycoprotein (GP). The black portions of the boxes represent the protein-coding portion of the gene and mRNA.
There is a subgroup of patients with SCLC and hyponatremia, however, who have no detectable levels of plasma AVP.32 Similar findings have been shown in SCLC cell lines.33 The tumors from these patients express ANP mRNA, secrete the peptide, and have high levels of ANP in their plasma.34 ANP is the leading candidate to be the natriuretic factor that Bartter and Schwartz proposed in their original description of SIADH. Further investigation into the precise role of ANP in patients with SCLC and hyponatremia of malignancy is ongoing.
 DIAGNOSIS
DIAGNOSIS
In patients with lung cancer, hyponatremia is most frequently diagnosed as a laboratory abnormality in the absence of significant symptoms. The symptoms of acute hyponatremia do not typically occur because the syndrome develops over a prolonged period in concert with the growth of the lung cancer. When symptomatic, mild hyponatremia (serum sodium <135 mmol/L) can cause headache, difficulty concentrating, nausea, weakness, and fatigue. Severe hyponatremia (serum sodium <125 mmol/L), especially when it develops rapidly, can lead to serious symptoms including confusion, hallucinations, seizures, coma, respiratory arrest, decerebrate posturing, and death.35
The diagnosis of SIADH is based on the following criteria: (1) plasma hypo-osmolality (<280 mOsm/kg); (2) osmolality of urine greater than serum (usually >500 mOsm/kg); (3) persistent urinary excretion of sodium in the absence of diuretics (>20 mEq/L); (4) absent signs of volume depletion; and (5) normal renal, adrenal, and thyroid function.36
In patients with lung cancer, nonmalignant causes of hyponatremia should be considered in the initial evaluation. These include diuretic use, renal disease, cardiac dysfunction, hypoadrenalism, thyroid disease, and dilutional hyponatremia. Medications as a cause for hyponatremia should also be addressed. Patients with lung cancer are commonly treated with cisplatin and narcotics, both of which can cause SIADH.
 TREATMENT
TREATMENT
The initial therapy for hyponatremia caused by lung cancer is treatment of the underlying malignancy. This often involves multimodality therapy with surgical excision, radiation, and/or chemotherapy. Even with tumor response to therapy, hyponatremia often persists or may recur following tumor progression. In SCLC, there are data to suggest that those patients who do not fully regain normal values of plasma sodium have a poorer survival than those patients who do.37,38
Asymptomatic patients with chronic hyponatremia are at low risk for serious neurologic sequelae, but are at risk of developing osmotic demyelination with rapid correction of the serum sodium concentration.39 Treatment is therefore aimed at gradual correction. Fluid restriction, which can be estimated by urinary and plasma electrolytes, but is usually started at 500 mL per day, is a cornerstone of therapy.40 Pharmacologic therapy may be indicated when fluid restriction is ineffective or poorly tolerated. Demeclocycline blocks the action of AVP on the renal tubule, thereby reducing urine osmolality and increasing serum sodium levels; however, its effects can be variable and it can cause nephrotoxicity.35 Lithium and phenytoin also inhibit the effects of AVP on the renal tubule, but are no longer recommended for treatment. A more recent option for the treatment of hyponatremia is vasopressin receptor antagonists. Conivaptan is a nonselective vasopressin receptor antagonist indicated for patients with moderate-to-severe hyponatremia with nonsevere symptoms and is administered intravenously.41 Tolvaptan is an orally active, selective AVP receptor 2 (V2) antagonist that has a similar indication. By blocking AVP effects in the renal collecting duct, aquaresis is promoted, leading to a controlled increase in serum sodium levels.42
For patients who present with severe, symptomatic hyponatremia characterized by seizure, delirium, or coma, rapid treatment is warranted.43 The widely accepted goal of therapy is to correct the serum sodium by 1 to 2 mmol/L per hour through the infusion of 3% saline. The administration of concomitant furosemide is also recommended. The magnitude of correction within the first 24 hours is suggested to be no more than 8 to 10 mmol/L and no more than 18 to 25 mmol/L in the first 48 hours to avoid osmotic demyelination. An alternative approach is to treat until acute symptoms resolve and then adjust the correction rate.44
ECTOPIC ADRENOCORTICOTROPIC HORMONE (ACTH) SYNDROME
Lung cancers are the most common neoplasms that cause ectopic ACTH production and Cushing syndrome, accounting for 50% of all cases. SCLC accounts for 80% to 90% of cases associated with lung cancer. Carcinoid tumors (10%) and bronchial adenocarcinomas (5%) have also been reported to produce biologically active ACTH. While biochemical abnormalities suggestive of ectopic ACTH production are reported in 30% to 50% of SCLC, Cushing syndrome is clinically present in only 1% to 5% of SCLC.45–50 Hypercortisolism is important to recognize as it increases the risk of therapy-induced complications51,52 such as opportunistic infections and venous thromboembolism (VTE).53–55
 BIOLOGY
BIOLOGY
Most cases of Cushing syndrome associated with lung cancers are caused by ectopic production of ACTH by the tumor.56 The precursor gene, proopiomelanocortin (POMC), is expressed in the cancer cells, and a 241-amino acid prohormone is translated and then cleaved into ACTH (39 amino acids), melanocyte-stimulating hormone, and opiate-like hormones (Fig. 118-3). The ACTH binds to receptors in the adrenal gland, causing them to produce excessive glucocorticoid and mineralocorticoid hormones.57
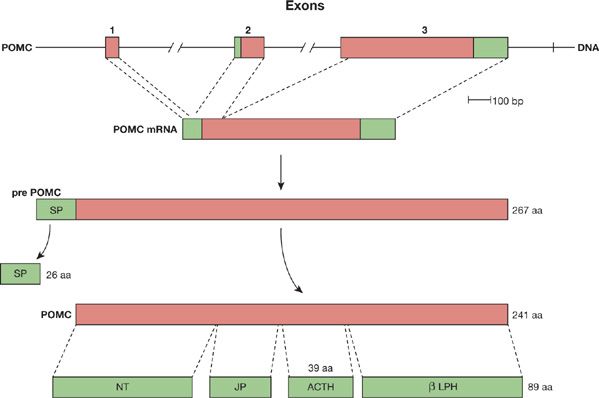
Figure 118-3 Proopiomelanocortin. The three exons of the POMC gene give rise to a 1072-base POMC mRNA. The mRNA is translated into a 267-amino acid pre-POMC with a 26-amino acid amino terminal signal peptide (SP). The SP is cleaved, creating the 241-amino acid POMC. The POMC peptide is cleaved into many products, including the N-terminal peptide (NT), the joining peptide (JP), a 39-amino acid mature ACTH, and β-lipotropin (β-LPH). The molecules can also undergo further processing. The black portions of the exons represent the protein-coding portion of the gene and mRNA.
Ectopic production of corticotropin-releasing hormone (CRH) has been reported as a cause of Cushing syndrome in a small number of patients with SCLC or bronchial carcinoids.49 CRH is a 41-amino acid peptide normally produced in the paraventricular nuclei of the hypothalamus that stimulates the release of ACTH from the pituitary.
 CLINICAL PRESENTATION AND DIAGNOSIS
CLINICAL PRESENTATION AND DIAGNOSIS
Ectopic ACTH production occurs with equal frequency in males and females, but Cushing syndrome has an 8:1 female preponderance. Patients who have slow-growing carcinoid tumors often present with the clinical features of Cushing syndrome: truncal obesity, moon facies, striae, polyuria, and polydipsia. In contrast, patients with SCLC often present with other signs of mineralocorticoid and glucocorticoid excess due to the rapidity of tumor growth: edema, weakness, hypertension, and hypokalemic alkalosis.
The diagnostic evaluation of ectopic ACTH syndrome starts by excluding iatrogenic Cushing syndrome in order to avoid unnecessary chemical testing. Once iatrogenic disease has been excluded, initial testing should include the following: 24-hour urinary-free cortisol, late-night salivary cortisol, and the low-dose dexamethasone suppression test.58–61 The diagnosis of Cushing syndrome is established when at least two of these tests are unequivocally abnormal. Demonstration of an increased plasma ACTH level (>22 pg/mL) confirms that the hypercortisolism is ACTH-dependent. Finally, persistently elevated serum cortisol in response to a high-dose dexamethasone suppression test differentiates ectopic ACTH syndrome from a pituitary source.58,62,63 Bronchial carcinoids are an exception, because in some tumors, ACTH and cortisol levels have been suppressed by dexamethasone.64,65
In patients with clinical features of Cushing syndrome in whom the dexamethasone suppression test is equivocal, a CRH stimulation test or bilateral inferior petrosal vein sampling will provide the definitive diagnosis. After CRH infusion, pituitary tumors release increased amounts of ACTH, whereas pituitary-independent lung tumors should not. Similarly, in pituitary-dependent Cushing syndrome, petrosal vein sampling will reveal a gradient between the level of ACTH in the petrosal vein and the peripheral concentration. In contrast, patients in whom ACTH is ectopically produced demonstrate no gradient between the petrosal vein and the peripheral blood.66,67
 TREATMENT
TREATMENT
Management of a patient with lung cancer and ectopic ACTH syndrome requires therapy directed at both the underlying tumor and the hypercortisolism. The treatment for a patient with ectopic ACTH production is to remove the source of the ACTH. This requires combination chemotherapy, with or without irradiation, for patients with SCLC and surgical resection and/or radiation for patients with carcinoid tumors. Chemotherapy for patients with SCLC and ectopic ACTH syndrome has been problematic. Patients often have a poor response to chemotherapy and are susceptible to early infection and death. Early control of a patient’s glucocorticoid excess is beneficial and may reduce the morbidity of treatment.
When removal of the ectopic source of ACTH is not possible, pharmacologic agents directed at blocking adrenal cortisol production may be successful. These drugs include ketoconazole, mitotane, metyrapone, and aminoglutethimide. Ketoconazole is an imidazole derivative that inhibits steroidogenesis at both adrenal and gonadal sites; it may be the most effective and least toxic agent available.68,69 Metapyrone and aminoglutethimide also have shown limited success by inhibiting adrenal steroid synthesis. Octreotide, a somatostatin analog, can suppress ectopic ACTH production and has been reported to be useful in some of these patients.68 Bilateral adrenalectomy should be considered in patients who fail medical therapy.62,70
In some patients, the clinical signs and symptoms of ectopic ACTH production develop before the development of a clinically obvious lung cancer. In these cases, symptomatic management of hypercortisolism is undertaken and periodic imaging studies are performed because these patients may have a slow-growing carcinoid tumor that will be amenable to surgical resection.64,71
ACROMEGALY
Only 1% of acromegaly is caused by ectopic production of growth hormone–releasing hormone (GHRH) or growth hormone (GH) by tumors.72 Of these, the majority are caused by carcinoid tumors of the lung and intestine.73
 BIOLOGY
BIOLOGY
The ectopic production of GHRH or GH by lung cancers has been demonstrated to cause acromegaly. In most cases, the GHRH gene is expressed by the cancer cells, and a 40- or 44-amino acid peptide is produced.74 Circulating GHRH peptide binds to receptors in the pituitary gland resulting in the production of excessive amounts of GH.75 GH then mediates its effects through GH receptors in soft tissue and by stimulating the production of insulin-like growth factor-1 (IGF-1).
Although many carcinoid tumors express immunoreactive GHRH and result in abnormal GH secretion,76–78 most patients with these tumors are not clinically acromegalic. It has been suggested that the observed high incidence of GHRH expression and low incidence of clinical acromegaly may be due to inadequate tumor production of GHRH or due to the impaired bioactivity of the circulating GHRH.75
 DIAGNOSIS
DIAGNOSIS
The earliest features of GH excess are hypertrophy of the extremities and face (often manifest as increased glove, shoe, and ring size), thickened leathery skin, prominent skin folds, hyperpigmented skin, and hair growth. Bony changes, hypertension, and diabetes mellitus are later, less common findings.
The presentation of a patient with a lung mass and signs of acromegaly should raise suspicion of ectopic acromegaly especially if the mass is found to be carcinoid. The diagnosis of ectopic acromegaly is established by elevated serum levels of GHRH or GH, the absence of a pituitary tumor, complete recovery following lung tumor resection, positive GHRH immunostaining, detection of GHRH mRNA, positive bioassay (cultured rat pituitary cells produce GH in response to tumor extract), or GHRH extraction from the tumor tissue.79 Because coincidental pituitary tumors and solid tumors have been described, patients who have lung cancer in association with low GHRH levels and high GH and IGF-1 levels should undergo magnetic resonance imaging (MRI) to exclude a pituitary tumor.
 TREATMENT
TREATMENT
Management of ectopic acromegaly involves surgical resection of the tumor and is often curative in those with lung carcinoid. In those with unresectable or metastatic cancers, medical therapy with somatostatin analogs, such as octreotide and bromocriptine, have been shown to be effective.80,81 Bromocriptine acts by inhibiting GH release by the pituitary; octreotide lowers both GH and IGF-1 levels in plasma and also appears to inhibit GHRH release by tumors. Clinical abatement of acromegalic features has been reported in patients treated with octreotide.
 CARCINOID SYNDROME
CARCINOID SYNDROME
Ectopic serotonin production is rare in lung neoplasms, occurring in 1% to 5% of patients with pulmonary neuroendocrine tumors (NETs).82 Hallmarks of the syndrome include skin flushing of the upper thorax, secretory diarrhea, and bronchoconstriction. It is important to recognize an acute carcinoid crisis in patients with lung cancer as it can be precipitated by chemotherapy, biopsy, anesthesia, surgery, or the use of adrenergic drugs. It is a result of a massive serotonin release, and can lead to cardiopulmonary failure.83
The evaluation for carcinoid syndrome begins with a 24-hour urine collection for the main metabolite of serotonin, 5-hydroxyindoleacetic acid (5-HIAA). This test has a specificity of approximately 90%.84 Detection of occult NETs in patients presenting with a carcinoid syndrome may be aided by testing for serum biomarkers of NETs such as neuron-specific enolase (NSE) and chromogranin A.85 When these serum biomarkers are negative in a patient with high clinical suspicion of a NET, radionuclide-labeled octreotide scintigraphy is useful as up to 80% of pulmonary NETs express somatostatin receptors.
In patients who are surgical candidates, surgery is the optimal treatment of carcinoid syndrome. Otherwise, control of symptoms should be the goal via somatostatin analogs, serotonin receptor blockers, interferon, and antidiarrheal medications. In addition, carcinoid crisis can be prevented or treated with intravenous octreotide acetate.83
HEMATOLOGIC SYNDROMES
Most hematologic syndromes associated with lung tumors are not as well characterized as the endocrine syndromes, and an ectopic hormone responsible for the syndrome has not been identified in most tumor tissues. In many of the hematologic syndromes, such as granulocytosis and thrombocytosis, clinical sequelae are often absent. As with the endocrine paraneoplastic syndromes, the most appropriate therapy for the hematologic syndromes is the treatment of the underlying neoplasm.
 GRANULOCYTOSIS
GRANULOCYTOSIS
NSCLC is the most common cancer associated with granulocytosis. Fifteen to twenty percent of patients with NSCLC have granulocytosis, with absolute white blood counts ranging from 10,100 to 25,000 (normal range is 4000–10,000). Its presence is associated with a poorer prognosis compared to patients without granulocytosis.86–88 Although granulocyte colony-stimulating activity can be demonstrated in serum and/or urine in 80% of affected patients, tumor production of granulocyte colony-stimulating factor, granulocyte monocyte colony-stimulating factor, or interleukin-6 (IL-6) has been shown in only a minority of patients.89,90
Virtually all patients with lung cancer who present with tumor-associated granulocytosis are asymptomatic. The diagnosis is suggested by the presence of an increased white blood count in which neutrophils predominate without immature forms, in the absence of non-neoplastic causes. An increased leukocyte alkaline phosphatase score and a normal bone marrow are consistent with this diagnosis.
 THROMBOCYTOSIS
THROMBOCYTOSIS
Thrombocytosis is common in patients with lung cancer, afflicting 16% to 32% of patients with both NSCLC and SCLC.91,92 The pathogenesis of thrombocytosis in patients with lung cancer has not been definitively elucidated. IL-6, which is a cytokine for megakaryocytes, has been demonstrated in cell lines from patients with lung cancer and thrombocytosis, and increased levels of IL-6 have been demonstrated in the plasma of such patients. Thrombopoietin is increased in patients with lung cancer and thrombocytosis but is decreased in patients with essential thrombocytosis. The identification of the thrombopoietin gene may lead to a better understanding of the role of this protein in paraneoplastic thrombocytosis.
Patients with thrombocytosis are nearly always asymptomatic and do not have an increased incidence of thromboembolism. The diagnosis of cancer-associated thrombocytosis is suggested by an increased platelet count (>500,000/mm2) in a patient with newly diagnosed lung cancer, and it is associated with advanced disease and worse clinical outcomes.93,94 A primary myeloproliferative disorder can be excluded by a bone marrow biopsy.95 In addition, the presence of the JAK2 V617F mutation may indicate essential thrombocytosis (present in 50% of cases) since it is not present in cases of reactive thrombocytosis.96
 THROMBOEMBOLISM
THROMBOEMBOLISM
Of all disease states, lung cancer is known to have one of the strongest associations with VTE.97 The incidence of VTE in patients with lung cancer is approximately 40 to 100 cases per 1000 person-years compared to 1 to 2 cases per 1000 person-years in the general population. It is also notable that 20% of patients who present with recurrent idiopathic venous thrombosis are found to have an underlying diagnosis of cancer.98 The spectrum of causes of thrombosis in patients with lung cancer is broad, including disseminated intravascular coagulation (DIC), Trousseau syndrome (recurrent migratory venous thrombophlebitis), nonbacterial thrombotic endocarditis, and obstruction of great vessels.99,100 Surgical procedures and chemotherapy have also been demonstrated to increase cancer patients’ risk of thrombotic complications.101
Cancer cells are able to directly activate the clotting cascade via two procoagulants: tissue factor (TF) and cancer procoagulant (CP). Increased expression of human TF has been shown in NSCLC tissue, and active TF-bearing microparticles, which may originate from tumor cells, have been demonstrated in the circulation of cancer patients.102,103 Microparticle-associated TF activity has been proposed as a mechanism for the prothrombotic state in cancer patients.104
The treatment of VTE associated with active lung cancer differs from standard treatment of VTE. It was previously shown that for recurrent thromboses, long-term subcutaneous heparin is more efficacious than warfarin.105 More recently, a meta-analysis of six randomized controlled trials in cancer patients being treated with long-term anticoagulation for VTE found no statistically significant survival benefit for low molecular weight heparin (LMWH) compared to warfarin, but a statistically significant reduction in VTE with LMWH, with no significant difference in bleeding.106,107
NEUROLOGIC SYNDROMES
There are a number of paraneoplastic neurologic syndromes associated with lung cancer. Encephalomyelitis, cerebellar degeneration, retinopathy, opsoclonus/myoclonus, and the Lambert–Eaton syndrome have all been described and are most commonly associated with SCLC. The majority of these syndromes are a result of an autoimmune response directed at shared antigens present in both cancer cells and normal neural tissue. The autoantibodies associated with each neurologic syndrome are listed in Table 118-2.



