Marfan syndrome (MS) is an inherited connective tissue disorder, and detailed evaluations of multiple organ systems are required for its diagnosis. Genetic testing of the disease-causing fibrillin-1 gene (FBN1) is also important in this diagnostic scheme. The aim of this study was to define the clinical characteristics of Japanese patients with MS and enable the efficient and accurate diagnosis of MS with mutational analysis using a high-throughput microarray-based resequencing system. Fifty-three Japanese probands were recruited, and their clinical characteristics were evaluated using the Ghent criteria. For mutational analysis, an oligonucleotide microarray was designed to interrogate FBN1, and the entire exon and exon-intron boundaries of FBN1 were sequenced. Clinical evaluation revealed more pulmonary phenotypes and fewer skeletal phenotypes in Japanese patients with MS compared to Caucasians. The microarray-based resequencing system detected 35 kinds of mutations, including 23 new mutations. The mutation detection rate for patients who fulfilled the Ghent criteria reached 71%. Of note, splicing mutations accounted for 19% of all mutations, which is more than previously reported. In conclusion, this comprehensive approach successfully detected clinical phenotypes of Japanese patients with MS and demonstrated the usefulness and feasibility of this microarray-based high-throughput resequencing system for mutational analysis of MS.
The Marfan syndrome (MS) is an inherited connective tissue disorder with an autosomal dominant inheritance, primarily involving the skeletal, ocular, and cardiovascular systems, caused by mutations in fibrillin-1 gene (FBN1). Diagnosis of the MS has been made using the Ghent criteria on the basis of data from European and American populations, but the Ghent criteria may not be completely suitable for the Japanese population. Therefore, epidemiologic and genetic surveys in the Japanese population are mandatory to establish more Japanese-specific (or Asian-specific) diagnostic criteria for the MS. The Ghent criteria were recently further revised. More weight is now given to FBN1 testing, and a diagnosis can be made if a patient has the FBN1 mutation plus either an aortic phenotype or ectopia lentis. These new criteria are much simpler than the original criteria. Thus, genetic testing of MS is becoming more important. FBN1 spans a 230-kb genomic region and contains 65 exons. More than 1,000 reported mutations are spread throughout the gene and are mostly unique in each affected family. Classic genetic analysis methods such as direct sequencing are very time consuming. Thus, the introduction of a more efficient genetic analysis tool is needed. Custom-designed resequencing microarrays enable the analysis of multiple genes spanning 30 to 300 kb on a single array. The microarray identifies individual nucleotides by comparative, high-fidelity hybridization using oligonucleotide probes ( Figure 1 ) . In the present study, we comprehensively evaluated the clinical characteristics of Japanese patients with suspected MS and also conducted mutational analysis of these patients by adopting a high-throughput genetic diagnosing system to achieve more efficient and accurate diagnoses.
Methods
Fifty-three consecutive probands suspected of having MS who visited the MS clinic at our hospital were enrolled. All patients were assessed using the original Ghent criteria. This study was conducted according to the Declaration of Helsinki and was approved by the institutional ethics committee. Written informed consent was obtained after providing a detailed explanation of the study. Genomic deoxyribonucleic acid (DNA) was extracted from buffy coat using a Genomix DNA extraction kit (Talent, Trieste, Italy). For amplification of the 65 exons of FBN1, polymerase chain reaction (PCR) primers were designed by referring to previous reports. After performing the PCRs according to the standard protocol, the PCR products were subjected to hybridization on the microarray.
The resequencing microarray was designed on the basis of the reference sequences from the Ensembl database. Because highly homologous sequences lead to cross-hybridization, FBN1 was checked for possible repetitive sequences using RepeatMasker software ( http://repeatmasker.org/chi-bin/webrepeatmasker ). No repetitive elements were observed. The microarray contained sense and antisense sequences for the 65 exons of FBN1 and ≥12 flanking base pairs of the splice junctions. The PCR product was fragmented, end-labeled with biotin, and hybridized to the array. Washing and staining with streptavidin-phycoerythrin were performed on automated fluidic stations according to the manufacturer’s protocol (Affymetrix, Santa Clara, California). Hybridization signals were read by a high-resolution laser scanner, and the data collection and interpretation were carried out using GeneChip Operating Software and GeneChip Sequence Analysis Software (Affymetrix), respectively.
Candidate nucleotide substitutions detected by the microarray-based resequencing system were subsequently validated by fluorescent dideoxy DNA sequencing using BigDye terminator version 3.1 on an ABI PRISM 3100xl genetic analyzer (Applied Biosystems, Foster City, California).
Some patients underwent cardiovascular surgery, and written informed consent for research use of surgical specimens was obtained from each patient. Total ribonucleic acid (RNA) was extracted using an RNeasy Fibrous Tissue Mini Kit (Qiagen, Venlo, The Netherlands). For patients whose aortic tissues were not available, total RNA was extracted from blood using a QIAamp RNA Blood Mini Kit (Qiagen). The RNA was converted to complementary DNA using SuperScript III First-Strand Synthesis SuperMix (Invitrogen, Carlsbad, California). PCR analyses were performed with specific primers designed for the target regions. PCR samples or subcloned plasmids after TA cloning of PCR products using a TOPO-TA vector (Invitrogen) were subjected to fluorescent dideoxy DNA sequencing.
DNA from patients whose mutations were not found by the aforementioned methods was screened by multiplex ligation-dependent probe amplification using a SALSA MLPA kit P065/P066 (MRC-Holland, Amsterdam, The Netherlands) for large deletions and duplications.
All quantitative data are expressed as mean ± SD. Statistical comparisons of distributions between groups were made using the chi-square test. Significance was taken as p <0.05.
Results
Of the 53 probands enrolled, 45 were diagnosed with MS according to the original Ghent criteria. Because our Marfan clinic offers cardiac surgery and some patients were referred for aortic surgery from other hospitals, most of the patients had aortic phenotypes ( Table 1 ). Dural ectasia and ectopia lentis were common findings, and positive family histories were seen in about half of the probands. We confirmed a lower frequency for some of the skeletal manifestations in Japanese patients with MS compared to that reported in a Western database, such as an arm span/height ratio >1.05 (20% in our study vs 55% in Western populations) and reduced extension at the elbows (<170°) (5% vs 15%), findings that were similar to the report of Akutsu et al ( Table 2 ). However, the frequency of major skeletal criteria (29%) was higher than a previous Japanese report (15%), which is partially due to a lack of evaluation of protrusio acetabuli in the earlier study. We found a higher frequency of spontaneous pneumothorax (30% vs 7%) in our Japanese population compared to a previous study conducted in Western patients. Calcification of the mitral annulus and frequency of dilatation of the main pulmonary artery were rarely reported. Actually, mitral annular calcification was not detected at all. However, pulmonary artery dilatation was relatively frequent (45% [9 of 20]) in our study, after excluding those patients whose main pulmonary artery diameters were difficult to evaluate.
| Variable | Total | Ghent-Positive Patients |
|---|---|---|
| (n = 53) | (n = 45) | |
| Age (years) | 33.1 ± 9.8 | 33.1 ± 10.4 |
| Men | 35/53 (66%) | 30/45 (67%) |
| Ghent positive | 45/53 (85%) | 45/45 (100%) |
| Skeletal major criteria | 12/49 (25%) | 12/41 (29%) |
| Skeletal minor criteria | 19/49 (39%) | 17/41 (42%) |
| Ectopia lentis | 25/53 (47%) | 25/45 (56%) |
| Cardiovascular major criteria | 48/53 (91%) | 44/45 (98%) |
| Cardiovascular minor criteria | 36/48 (75%) | 32/41 (78%) |
| Pulmonary | 22/49 (45%) | 19/42 (45%) |
| Skin | 26/49 (53%) | 23/42 (55%) |
| Dural ectasia | 34/47 (72%) | 33/40 (83%) |
| Family history of MS | 31/55 (56%) | 28/45 (62%) |
| Criterion | n (%) |
|---|---|
| Skeletal major criteria | |
| Pectus carinatum | 9/42 (21%) |
| Pectus excavatum, requiring surgery | 7/44 (16%) |
| Arm span/height ratio >1.05 | 8/41 (20%) |
| Wrist and thumb signs | 32/43 (74%) |
| Scoliosis of >20% or spondylolisthesis | 21/44 (48%) |
| Reduced extension at the elbows (<170°) | 2/41 (5%) |
| Medial displacement of medial malleolus, causing pes planus | 16/41 (39%) |
| Protrusio acetabuli | 8/39 (21%) |
| Skeletal minor criteria | |
| Pectus excavatum of moderate severity | 10/44 (23%) |
| Joint hypermobility | 7/41 (17%) |
| Highly arched palate with crowding of teeth | 31/40 (78%) |
| Facial appearance | 15/40 (38%) |
| Cardiovascular major criteria | |
| Dilatation/dissection of the ascending aorta | 44/45 (98%) |
| Cardiovascular minor criteria | |
| Mitral valve prolapse | 23/42 (55%) |
| Dilatation of main pulmonary artery | 9/20 (45%) |
| Calcification of mitral annulus | 0/34 (0%) |
| Dilatation/dissection of descending thoracic/abdominal aorta | 12/43 (28%) |
| Pulmonary minor criteria | |
| Spontaneous pneumothorax | 13/43 (30%) |
| Apical blebs | 15/44 (34%) |
| Skin minor criteria | |
| Striae atrophicae | 24/42 (57%) |
| Recurrent or incisional herniae | 0/41 (0%) |
In our mutational analysis, the base call rate of this system for FBN1 was >96% when examining 5 representative cases, and resequencing as many as 12,688 bp per patient was easily accomplished in 3 working days, demonstrating the high fidelity and high throughput of this system.
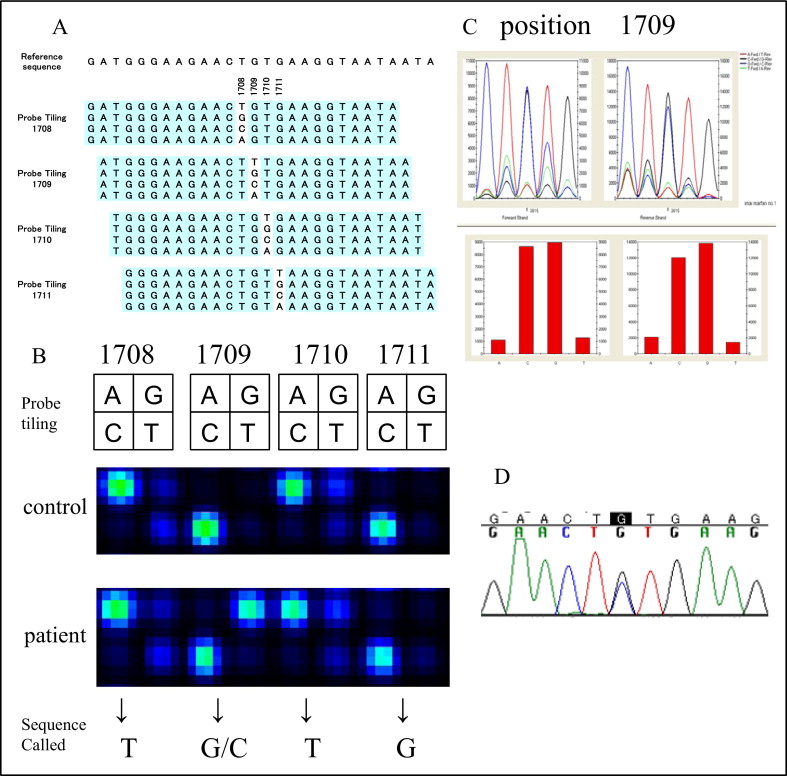
In the 53 probands, 35 kinds of FBN1 mutations were found in 36 probands using this system ( Table 3 ). There were 18 missense and 9 nonsense mutations. Eight other mutations located near the exon-intron boundaries were thought to alter the splicing patterns. Supplemental direct sequencing in probands with no mutation detected by the microarray-based method revealed 5 deletion mutations in FBN1 ( Table 3 ). Furthermore, multiplex ligation-dependent probe amplification assay revealed a large deletion mutation (exon 3) in 1 proband. Finally, novel mutations were found in 23 probands using microarray and in 29 probands in total. All possible mutations found by the microarray-based resequencing system were verified by direct sequencing, and thus the microarray detected point mutations with 100% accuracy. A representative example of genetic analysis using the microarray-based resequencing system is shown in Figure 1 . Of 18 missense mutations, 11 were either affecting or creating cysteine residues. For other novel missense mutations, none of the mutations were found in ≥200 ethnically matched control subjects. The mutation detection rate of the microarray-based resequencing system for the Ghent-positive patients was 71%. The overall mutation detection rate after additional analysis by fluorescent dideoxy DNA sequencing and multiplex ligation-dependent probe amplification reached 84% ( Table 4 ).
| Exon | Complementary DNA | Protein |
|---|---|---|
| Missense mutations | ||
| 4 | c.386G>A | p.Cys 129 Tyr |
| 13 | c.1709G>C ⁎ | p.Cys 570 Ser |
| 14 | c.1786T>G ⁎ | p.Cys 596 Gly |
| 15 | c.1911T>G ⁎ | p.Cys 637 Trp |
| 18 | c.2171T>G ⁎ | p.Ile 724 Arg |
| 18 | c.2201G>T | p.Cys 734 Phe |
| 21 | c.2638G>A | p.Gly 880 Ser |
| 24 | c.3043G>A | p.Ala 1015 Thr |
| 26 | c.3263A>G ⁎ | p.Asn 1088 Ser |
| 28 | c.3503A>G | p.Asn 1168 Ser |
| 34 | c.4280A>G ⁎ | p.Tyr 1427 Cys |
| 43 | c.5371T>C ⁎ | p.Cys 1791 Arg |
| 47 | c.5873G>A ⁎ | p.Cys 1958 Tyr |
| 50 | c.6296G>T | p.Cys 2099 Phe |
| 53 | c.6518G>A ⁎ | p.Gly 2173 Ser |
| 57 | c.7015T>G ⁎ | p.Cys 2339 Gly |
| 60 | c.7466G>A ⁎ | p.Cys 2489 Tyr |
| 62 | c.7754T>C | p.Ile 2585 Thr (2 probands) |
| Nonsense mutations | ||
| 8 | c.945T>A ⁎ | p.Cys 315 X |
| 12 | c.1585C>T | p.Arg 529 X |
| 29 | c.3603C>A ⁎ | p.Cys 1201 X |
| 37 | c.4709G>A ⁎ | p.Trp 1570 X |
| 38 | c.4777G>T ⁎ | p.Glu 1593 X |
| 38 | c.4786C>T | p.Arg 1596 X |
| 54 | c.6658C>T | p.Arg 2220 X |
| 58 | c.7240C>T | p.Arg 2414 X |
| 65 | c.8521G>T ⁎ | p.Glu 2841 X |
| Splicing mutations | ||
| 11–12 | c.IVS11+5G>A | p.Cys474Tyr Glu475_Asp490del |
| 15–16 | c.IVS15-3T>G ⁎ | |
| 16–17 | c.IVS16+3A>C ⁎ | |
| 18–19 | c.IVS18+1G>C ⁎ | |
| 34–35 | c.IVS34-1G>A ⁎ | p.Asp1446ValfsX21 |
| 40–41 | c.IVS40+1G>A ⁎ | |
| 52–53 | c.6453C>T ⁎ | p.Cys2151Tyr, Glu2152_Asp2166del |
| 56–57 | c.IVS56+5G>A ⁎ | |
| Deletion mutations | ||
| 54 | c.6665delT ⁎ | p.Val2222GlyFsX69 |
| 54 | c.6703-6704delGG ⁎ | p.Gly2235IlefsX7 |
| 55 | c.6837delG ⁎ | p.Tyr2280IlefsX10 |
| 57 | c. 7071_7079delCGTCACCAA ⁎ | p.Val2358SerfsX511 |
| 65 | c. 8532_8delTACAACT ⁎ | p.Thr2785X |
| 3 | Exon 3 deletion ⁎ |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


