Epidemiology and Prevention of Congenital Heart Defects
Lorenzo D. Botto
From Epidemiology to Prevention
Few questions are as poignant as those that many parents pose after finding out that their baby has a serious heart defect—“what will happen to my child?” (outcome), “why did this happen?” (causation), and, at some point, “what can we do so it will not happen again?” (prevention). In different guises, these questions continue to challenge clinicians and public health professionals. Remarkably, today’s answers are typically partial, fragmentary, and based on evidence that is rarely current, validated, and population-based. Also, integrating the various strands of evidence in a lifespan, person-centered thread remains a goal far from completion.
Epidemiology can help fill these gaps. With their focus on methodologically sound evaluation of health metrics in human populations, epidemiologic studies can complement the wealth of clinical data originating from descriptive case series and reports, and shed light (or raise questions) on the evidence relating to outcomes, causation, and prevention of congenital heart defects. The focus of the chapter is not only to summarize current data but also to highlight the more urgent gaps and discuss trends that are likely to shape the future. The discussion is necessarily selective and draws not only on primary literature but also on several reviews, where additional information can be found (1,2,3,4,5).
Epidemiology, at its core, is a practical science: it measures in order to generate data for action. In this context, data for action includes information targeted to raising awareness of the massive burden of disease associated with congenital heart defects (incentive for change), characterizing modifiable determinants of disease (evidence for change), and, once interventions are deployed, assessing the effectiveness of prevention strategies (Fig. 2.1).
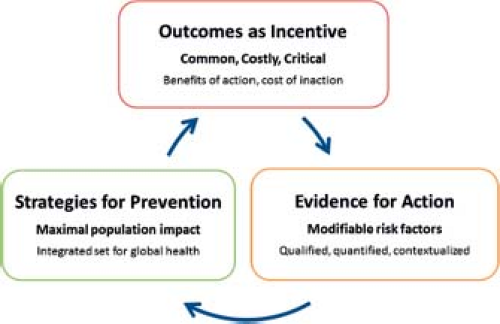 Figure 2.1 The cycle from epidemiology to prevention: incentive, evidence, strategies. |
Specifically, the “descriptive epidemiology” of congenital heart defects—frequency and outcomes—provides crucial measures of the impact on individuals and society and represents materially the incentive for change. Such metrics also helps convey the human cost of inaction—lost lives, illness and disability, wasted monies—if best practices in treatment and prevention are not implemented and improved. In this sense, the descriptive epidemiology also provides a policy and moral basis for prevention.
The “analytic epidemiology” of congenital heart defects—epidemiology studies aimed at characterizing modifiable causes in human population—provides the actionable evidence required for interventions. Such information on impact and causation can be filtered through the lens of the attributable fraction of different risk factors in a population (estimating the relative contribution of these risk factors to the burden of disease in a specific population); in conjunction with an understanding of the effectiveness of specific interventions, these data can help develop strategies aimed at the greatest benefit within the constraints of a country’s limited resources. Finally, the effects of the interventions need to be measured—this new assessment, through a new round of “descriptive epidemiology” becomes the new baseline and benchmark, in a continuous cycle of evidence-based health improvement and promotion. These three linked issues—outcome assessment, evidence on modifiable factors, and strategies for prevention—will be briefly discussed in turn.
Outcomes as Incentive for Action: Common, Costly, and Critical
Congenital heart defects are common, costly, critical—epidemiology supports this—and they are more so than they ought to be—because even causes that are known are not effectively prevented. As a basis for action, the evidence about outcome must be reliable (valid and possibly precise), recent (or at least timely), and relevant (to the targeted population and interventions). Remarkably, these standards are rarely, if ever, met—the data are often fragmentary (if present at all), inconsistent, or old. Nevertheless, it is possible to sketch a provisional answer to the three main questions related to the impact of congenital heart defects—how common, how costly, and how critical.
How Common are Congenital Heart Defects?
Overall, the birth prevalence of major congenital heart defects is approximately 1% (2,5,6,7,8,9,10,11). Estimating this figure is the subject of studies, reviews, and debates (5,6,9,11). The summary estimate is particularly important for raising awareness. For most other practical purposes, however, it is nearly useless and possibly misleading. Because of their heterogeneity in clinical
presentation, health impact, and, to a degree, risk factor profile, it is nearly always more useful to evaluate subsets of congenital heart defects (grouped by anatomy, severity, or embryology, depending on the goal of the analysis), rather than all heart defects combined. For example, the prevalence, time trends, and inter-program variability vary significantly (Fig. 2.2), and the same can be said about health outcomes and cost.
presentation, health impact, and, to a degree, risk factor profile, it is nearly always more useful to evaluate subsets of congenital heart defects (grouped by anatomy, severity, or embryology, depending on the goal of the analysis), rather than all heart defects combined. For example, the prevalence, time trends, and inter-program variability vary significantly (Fig. 2.2), and the same can be said about health outcomes and cost.
A subset of more severe conditions are fewer in number and their reported prevalence shows limited variability across studies. As a group, they occur in 2 to 3 per 1,000 births. What constitutes a “severe” heart defect varies somewhat between studies (12,13,14), but the overall finding remains the same. This relatively small group of conditions contributes to a disproportionately high fraction of death, disability, and cost associated with congenital heart defects. The remaining group—mostly septal defects, pulmonic and aortic stenosis—are usually (but not always) clinically milder and more common (Fig. 2.2), occurring in 4 to 8 per 1,000 births. Their birth prevalence is much more variable across studies, and because they are so frequent, these conditions drive most of the reported variation across studies of the overall frequency of heart defects (between 5 and 15 per 1000 in this example) (5,11,15,16,17,18). International surveys and comprehensive meta-analyses reach these same broad conclusions (6,7,9). Such variability would be even greater, if some mild conditions such as small muscular ventricular septal defects are included—these latter conditions, nearly always clinically benign and often resolving spontaneously, may be found in 1% or more newborns if systematically scanned by echocardiogram (18,19,20).
Rates can then be translated into numbers of babies born in a population in a given time period (3,10,11). This metric provides a measure of the frequency of congenital heart defects that can be more directly helpful in healthcare planning and policy development. Applying the rate estimates just discussed—and with the caveat that rates in developed countries may not necessarily applicable worldwide—leads (Fig. 2.3) to an estimated 1.2 million affected babies born every year worldwide (9 per 1,000 of 135 million births). Of these, at least 300,000 (2 to 3 per 1,000) will have severe conditions that require prompt, and often complex surgical and medical care to avoid death or disability.
These estimates also underscore how most affected babies will be born in countries with few of the resources needed for optimal treatment and management (Fig. 2.4). In all countries, but particularly these, primary prevention, costing less, would be particularly beneficial.
What Drives the Variations in Reported Birth Prevalence?
The variation in birth prevalence of the milder defects (which drive the overall rate) has never been convincingly proven to be real. The several-fold increase in these rates in recent decades, concomitant with the diffusion of echocardiography, supports the primary role of methodology, rather than biology, in driving these changes (5,9,21,22,23). The time trend of right obstructive defects observed in a population-based
US program (21) provides a telling illustration of this point (Fig. 2.5), and is only one of several possible examples. For example, ventricular septal defects show similar trends in many programs (5).
US program (21) provides a telling illustration of this point (Fig. 2.5), and is only one of several possible examples. For example, ventricular septal defects show similar trends in many programs (5).
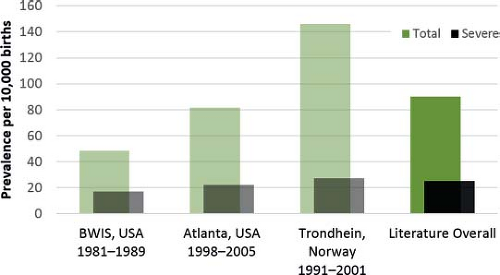 Figure 2.2 Birth prevalence of major congenital heart defects (overall and severe) in three studies compared to estimated averages from the literature (overall rate, 9 per 1,000; rate for severe heart defects, 2.5 per 1,000). Note: Severe heart defects include single ventricle, heterotaxy, conotruncal defects, atrioventricular septal defects, total anomalous venous return, hypoplastic left heart, coarctation of the aorta, interrupted aortic arch, and pulmonary atresia. Total major heart defects exclude bicuspid aortic valve and patent ductus arteriosus. (From Ferencz C, Loffredo CA, Rubin JD, Magee CA. Epidemiology of congenital heart disease: the Baltimore-Washington Infant Study 1981–1989. Mount Kisco, NY: Futura Publishing Company, Inc.; 1993; Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–1900; Tegnander E, Williams W, Johansen OJ, Blaas HG, Eik-Nes SH. Prenatal detection of heart defects in a non-selected population of 30,149 fetuses—detection rates and outcome. Ultrasound Obstet Gynecol. 2006;27(3):252–265; Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr. 2008;153(6):807–813; van der Linde D, Konings EE, Slager MA, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58(21):2241–2247.) |
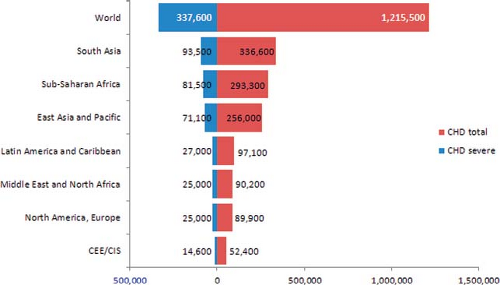 Figure 2.3 Estimated number of babies born yearly in different regions of the world with major heart defects (birth prevalence, 9 per 1,000) and with severe heart defects (2.5 per 1,000). (From Botto LD. Epidemiology and prevention of congenital heart defects. In: Muenke M, Kruszka PS, Sable CA, Belmont JW, eds. Congenital Heart Disease: Molecular Genetics, Principles of Diagnosis and Treatment: S. Karger AG, Basel, 2015.) |
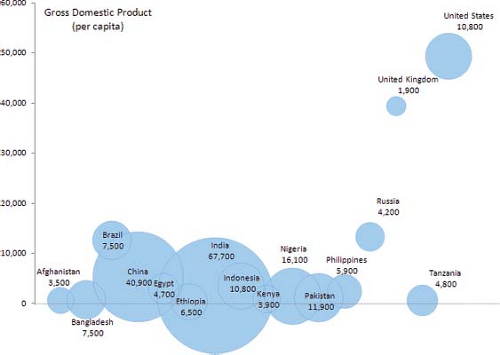 Figure 2.4 Estimated number of babies born with severe congenital heart defects (birth prevalence, 2.5 per 1,000) in selected countries, by per capita gross domestic product. The countries represented account for over 60% of all births worldwide. Area of the circles are proportional to the number of affected babies. Data from UNICEF and World Health Organization, 2011. (From Botto LD. Epidemiology and prevention of congenital heart defects. In: Muenke M, Kruszka PS, Sable CA, Belmont JW, eds. Congenital Heart Disease: Molecular Genetics, Principles of Diagnosis and Treatment: S. Karger AG, Basel, 2015.) |
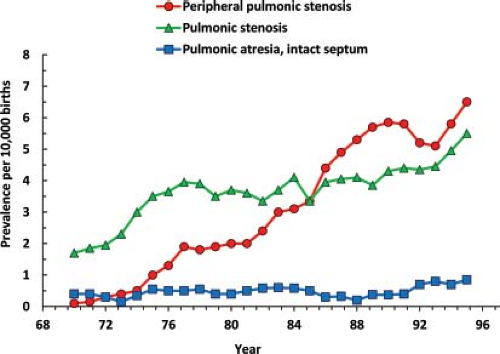 Figure 2.5 Time trends in the birth prevalence of selected right obstructive heart defects, Metropolitan Atlanta Congenital Defects Program, US. (From Botto LD, Correa A, Erickson JD. Racial and temporal variations in the prevalence of heart defects. Pediatrics. 2001;107(3):E32.) |
Among the right-sided obstructive defects, reported rates increased much more for the milder than the more severe defects—rising sharply and to greater heights for peripheral pulmonic stenosis, less so for valvar stenosis, and minimally, if at all, for pulmonary atresia with intact septum (21). These and other data underscore the importance of evaluating the many methodologic factors that can raise or lower reported rates—for example, the relatively recent decline in some congenital heart defects in Europe (24). Particular mention deserve the processes and quality standards related to case ascertainment, reporting, review, coding, and classification (2,6,7,25).
Additional factors include the reporting and inclusion of fetal deaths (26,27,28), and, particularly in recent years, of pregnancy terminations. The example of hypoplastic left heart syndrome is telling: The fraction of cases prenatally diagnosed and terminated varies greatly in countries and can exceed 50% (2,29,30). Small changes in how these cases are reported can influence rates considerably; also, not capturing these cases will underestimate, sometimes to a large degree, the burden of congenital heart defects.
Not all variation is necessarily spurious. Small differences in birth prevalence by sex or ethnic origin are probably real. Several studies have indicated slightly higher rates of d-transposition of the great arteries and left-sided obstructive defects in boys than in girls, and in non-Hispanic whites than in non-Hispanic blacks (17,21,31,32,33,34); these variations have yet to be satisfactorily explained in terms of either biology or methodology.
How Many People are Living with Congenital Heart Defects—Including Adults?
Counting how many people are living with congenital heart defects in the general population has proven remarkably difficult. Surprisingly few primary data are available. A major challenge is the general lack of accurate, population-based sources of data with validated diagnoses. Instead, available databases are typically dispersed across diverse health delivery systems and service providers, are designed for purposes other than clinical or public health evaluation, and include diagnoses based on administrative coding. In the absence of well-designed national surveillance activities for heart defects for adolescents and adults, monitoring prevalence and outcomes beyond childhood has proven extremely challenging. Currently, direct, population-based information on lifetime population prevalence is scarce. Most of what is available comes from a few areas or countries (35), and often relies either on statistical modeling (12) or linkages between administrative databases (14,36).
Yet, even these preliminary estimates highlight relatively consistent and important findings and trends. First, numbers are high: nearly 1 in every 200 people in the United States was estimated to be living with a heart defect (12), and nearly 1 in 160 adults in Quebec (14,36). A recent review of the literature focusing on adults only (35) identified ten reports from Europe, Japan, and North America and suggested an overall population prevalence of about 1 in 330 adults (3,000 per million population). Second, in both areas (12,14), there were more adults than children living with heart defects—two adults for every child, in one recent estimate from Quebec (14), reflecting the increasing life expectancy in recent decades. Third, these numbers are trending upward (12,36), by about 5% per year in one estimate (37)—in Quebec, the estimated prevalence among adults increased by over 50% between 2000 and 2010, and included many severe congenital heart defects than previously reported (14). These findings, despite their limitations, support the urgency of investing in the specialized care of adolescents and adults with congenital heart defects. They also underscore the need for systematic, population-based surveillance programs that can track over time the evolution of these trends through the lifespan, not only children, as these trends have obvious implications for quality of care, health services planning, and cost.
How Costly are Congenital Heart Defects?
Cost and use of resources are a growing area of concern and interest, and rightly so. Congenital heart defects are the most expensive of all birth defects (38,39). In the United States, six of the ten birth defects with highest hospital charges were congenital heart defects, and two—hypoplastic left heart and truncus arteriosus—topped the list (38). In a large survey of children’s hospitals, nine types of congenital heart defects were in the top tier of cost (39). Costs can increase markedly in the presence of complications, and longer length of stay (40).
Whereas such research throughput is increasing, generating comprehensive estimates of cost continue to be extremely challenging. Not only do costs vary by institution (41), but in general depend on context, perspective, and definitions. The local context is critical: Clinical factors (survival, rate of complications) and organizational systems (health delivery systems, payor structure) vary
by country, within country sometimes, and over time, in directions that are not always obvious or predictable. Thus, cost estimates cannot be simply “borrowed” from one country to the next. Cost estimates also depend on perspective: depending on whose view is taken—the patient (client), the health delivery system, or society as a whole—estimates can vary considerably. Finally, definitions, and in particular inclusion criteria, are crucial: inpatient medical costs, for which data are comparatively easy to obtain, are an important component of cost, but not the only one; other direct and indirect costs, including loss of productivity, can be considerable and may exceed the inpatient costs, but are more difficult to estimate. A further challenge is estimating costs over the lifespan, which would provide a realistic assessment of the benefits of prevention. The complexities of cost estimation are many (42,43,44). Here only a few points are summarized (Fig. 2.6).
by country, within country sometimes, and over time, in directions that are not always obvious or predictable. Thus, cost estimates cannot be simply “borrowed” from one country to the next. Cost estimates also depend on perspective: depending on whose view is taken—the patient (client), the health delivery system, or society as a whole—estimates can vary considerably. Finally, definitions, and in particular inclusion criteria, are crucial: inpatient medical costs, for which data are comparatively easy to obtain, are an important component of cost, but not the only one; other direct and indirect costs, including loss of productivity, can be considerable and may exceed the inpatient costs, but are more difficult to estimate. A further challenge is estimating costs over the lifespan, which would provide a realistic assessment of the benefits of prevention. The complexities of cost estimation are many (42,43,44). Here only a few points are summarized (Fig. 2.6).
Short-Term Assessment: Cross-Sectional Costs
Cross-sectional costs are more readily available and can provide an immediate if rough estimate of the potential impact of prevention on cost. One study estimated an 1.4 billion dollars as the cost in a single year (2004) for inpatient admissions in the United States of people with congenital heart defects (42,45). Over one-third of this figure, 511 million dollars, was due to a small subset of severe heart defects—conotruncal defects, single ventricle, hypoplastic left heart syndrome, Ebstein anomaly, and atrioventricular septal defects. Infants accounted for 95% of these inpatient costs. In an attempt to incorporate outpatient data also, researchers used a different dataset limited to a privately insured population (33), and estimated medical costs (inpatient and outpatient) associated with major heart defects to be approximately $100,000 among children up to 3 years of age. For adults, few data are available, and these are likely underestimates due to the uncertainties of coding in administrative datasets (46).
Long-Term View: Lifetime Costs
Compared to cross-sectional costs in a given age range, lifetime estimates can provide a more accurate view of the benefits of prevention—for example, preventing a diabetes-associated heart defect saves costs over a lifetime of that baby. Because of this longitudinal component, estimating lifetime costs is understandably challenging, and requires modeling, data, and assumptions. Comprehensive lifetime estimates are few. An older study, but still one of the more comprehensive, estimated lifetime costs of 1.2 billion (in 1992 dollars) for a single year birth cohort for children born in the United States with one of four congenital heart defects—truncus arteriosus, d-transposition of the great arteries, tetralogy of Fallot, and single ventricle. Of these, direct medical costs, mainly surgical, were approximately 0.5 billion dollars (43,44); indirect costs, including loss of productivity, were the greater part of the total estimate.
Even from this brief review, it is evident that comprehensive costs estimates are remarkably scarce, surprisingly so in an environment of increasing healthcare expenditures and limited resources. With few exceptions, available estimates are also far from timely, a major limitation for such a dynamic issue as cost. These gaps reflect in part the limitations and accessibility of current data sources, often fragmented and opaque. Greater transparency and integration will be required for reliable cost information.
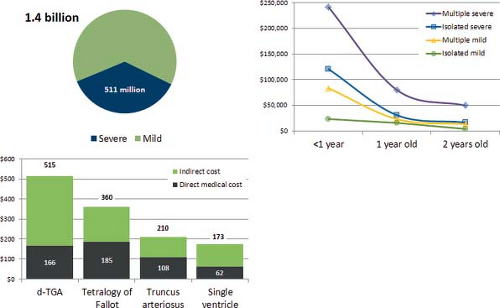 Figure 2.6 Estimates of costs for congenital heart defects, selected data. Top Left: Estimated inpatient cost for 1 year due to congenital heart defects (overall and for severe types) in 2004, United States. Top Right: Inpatient and outpatient costs (Mediscan data) for children under 3 years of age, by type of congenital heart defect, United States. Bottom Left: Estimated lifetime costs (direct and indirect, in 1992 dollars) per case, United States. (From Waitzman NJ, Romano PS, Scheffler RM. The Cost Of Birth Defects. University Press of America; 1996; Boulet S, Grosse SC, Riehle-Colarusso T, Correa-Villasenor A. Health care costs of congenital heart defects. In: Wyszynski DF, Correa-Villasenor A, Graham TP, eds. Congenital Heart Defects: From Origin To Treatment. New York, NY: Oxford University Press; 2010:493–501; Botto LD. Epidemiology and Prevention of Congenital Heart Defects. In: Muenke M, Kruszka PS, Sable CA, Belmont JW, eds. Congenital Heart Disease: Molecular Genetics, Principles of Diagnosis and Treatment: S. Karger AG, Basel, 2015.) |
How Critical are Heart Defects?
Much like cost, health outcomes—mortality, morbidity, disability, quality of life—are best evaluated within one’s local setting. Evaluating and tracking outcomes locally can help identify population-specific problems and disparities and their local determinants. Despite massive variations, some broad qualitative considerations can be made. Nearly everywhere, congenital heart defects are significant contributors to adverse health outcomes (2).
Mortality
Internationally, congenital heart defects are the leading cause of infant deaths due to congenital anomalies—accounting for approximately 1 in 3 such infant deaths (47,48). The contribution to neonatal deaths is also significant—in the United States (49) and in several European countries (13) congenital heart defects account for an estimated 1 in 4 neonatal deaths to birth defects. In developed countries, congenital heart defects are estimated to account for approximately 1 in 10 infant deaths from any cause (47,48).
In infants and young children, a disproportionate fraction of deaths is due to relatively few types of heart defects, particularly hypoplastic left heart syndrome, conotruncal defects, and atrioventricular septal defects (50). This finding further underscores the importance of monitoring, preventing, and treating the subset of severe congenital heart defects. Notably, excess mortality does not end in early childhood, but extends for many decades in adult life, as documented in a longitudinal population-based study in Denmark (51) as well as in a study of death certificates in the United States (52).
Developmental Disabilities
Adverse neurodevelopmental outcomes are an increasingly appreciated component of the burden of disease for people with congenital heart defects. In one survey, clinicians and parents of children with congenital heart defects rated neurologic disability a greater concern than cardiac disability (53). With longer life expectancy, these outcomes are increasingly relevant (54,55,56,57,58,59,60,61,62,63,64).
Neurodevelopmental outcomes are discussed in detail elsewhere (see Chapter 74). From an epidemiologic perspective, several points can be made. First, neurodevelopmental challenges are common when additional malformations or a genetic syndrome is present—even when a genetic condition is suspected but not specifically identified (65). Brain anomalies, such as neuronal migration defects and Chiari I malformation can be found in children with apparently isolated congenital heart defects (66). Fetal brain changes have been reported in some children with congenital heart defects (61,67), on the basis of volumetric analysis of the fetal brain and on magnetic resonance spectroscopy (67). After birth, suggested risk factors for adverse neurodevelopmental outcomes include altered hemodynamics, cyanosis, or the stress related to complications of surgery, low birth weight, or preterm birth (54,62,65,68,69,70,71,72,73). In general, intellectual disability is likely rare in the absence of genetic conditions and severe postnatal complications. However, whereas overall intelligence is typically within the normal range in older children and adolescents, other subtler findings—deficits in executive function, attention deficit and hyperactivity disorders, anxiety, and depression—could be more common than previously thought. Reviews suggest that a significant fraction of children and young adults who underwent open-heart surgery for CHD were at risk for adverse neurodevelopmental outcomes if not necessarily overt intellectual disability (68,74). Several investigators have voiced methodologic concerns in current studies—including the general moderate quality of the studies (106) and the scarcity of longitudinal studies (with most studies being cross-sectional). Longitudinal, prospective studies are difficult, but possible. In the Norwegian Mother and Child Cohort Study (MoBa), investigators linked the cohort of 44,000 children to the Norwegian nationwide heart defect registry and identified 175 children of 3 years of age with congenital heart defects, 60 of whom had severe defects. They then linked back to maternal questionnaires relative to the child’s motor, communication, and social impairment that were collected prospectively at birth, 6, 18, and 36 months. Compared to controls, children with severe heart defects had more than a three-fold risk for communication and gross motor impairments, and a two-fold increased risk for any developmental impairment. Children with mild and moderate heart defects had a two-fold higher risk for gross motor impairment but did not otherwise differ from controls. Of note, impairments were more frequent among children already noted to have developmental delays and a smaller head size at birth (75). The investigators recommended early assessment for motor and communication support provided in children with congenital heart defects, particularly severe types, with the goal of improving long-term outcomes. Notably, these same children did not have a higher risk of internalizing or externalizing emotional problems at 36 months of age compared to controls (76), possibly because they were past the main period of morbidity and hospitalizations.
These studies are notable for their systematic, longitudinal, and prospective design, and will hopefully continue to generate important outcome data on older children and adults, with fewer biases and greater generalizability than clinic-based case series on which most current data derives. In summary, some children with congenital heart defects appear to be at risk for adverse development and psychological outcomes, although firm data on frequency, magnitude of risk, and predictors are still scarce. Also, incorporating these outcomes into the “cost” of congenital heart defects would provide a more realistic evaluation of the potential benefits of primary prevention, and a further incentive for research and preventive interventions.
Quality of Life
Together with neurodevelopment, health-related quality of life is increasingly and appropriately viewed as a significant outcome in pediatric cardiology (see Chapter 77) (58,77,78). Quality of life has been defined as a multidimensional construct that integrates an individual’s subjective perception of physical, social, emotional, and cognitive functioning. Crucially, this construct is driven by the perspective of the person with a congenital heart defect (and the family), and integrates domains such as school functioning, social functioning, and independent living (56). For these reasons, quality of life metrics provide a view that is often missed when focusing exclusively on clinic data or administrative records, and require from investigators new skill sets, novel methods, and specifically designed tools (79,80,81,82). In fact, the scarcity of validated assessment tools targeted at people with congenital heart defects and their families have been a major challenge. Two recent reviews of the literature have highlighted the heterogeneity of methods, definitions, and assessment tools in quality of life studies in people with congenital heart defects, as well as the limited quality of some reports (74,78). These limitations add to the challenges of combining and understanding the aggregate data, particular in the presence of inconsistent or unexpected results (78).
A further challenge is that findings in one country or population may not be directly transferable elsewhere—precisely because quality of life is expected to depend not only on medical issues (e.g., disease severity, type of surgery) but also on healthcare system, family support, income, and societal attitudes toward chronic illness (58,83,84,85,86,87).
The overall conclusion in the reviewed data seems to be that the quality of life of adult patients with congenital heart disease appears to be compromised in physical domains, possibly less so in the psychosocial domains; however, results are quite variable, among adults and in children, as well as between children and their parents (74,78). For example, a population-based study in Finland found
reasonably good outcomes (educational attainment, employment level, and frequency of steady relationship) in a group of people with mild to moderate congenital heart defects (88). By contrast, several studies in North America and Europe reported worse health-related quality of life in people with congenital heart defects compared to reference groups (89,90,91,92). In some studies, these outcomes varied by anatomic lesion and surgeries (85,93), family income (83), and age (94,95,96). In the United States, obtaining employment, health insurance, and mortgages were noted as challenges in the United States (97), even for people with mild heart defects (98).
reasonably good outcomes (educational attainment, employment level, and frequency of steady relationship) in a group of people with mild to moderate congenital heart defects (88). By contrast, several studies in North America and Europe reported worse health-related quality of life in people with congenital heart defects compared to reference groups (89,90,91,92). In some studies, these outcomes varied by anatomic lesion and surgeries (85,93), family income (83), and age (94,95,96). In the United States, obtaining employment, health insurance, and mortgages were noted as challenges in the United States (97), even for people with mild heart defects (98).
Unsurprisingly, given the complexity of the issues and the variability of methods and focus, identifying simple or common predictors of quality of life has proven difficult. Only in a minority of studies quality of life was associated with the complexity of heart defects, type of surgery, duration of circulatory arrest, and number of surgical procedures (74). Caretaker issues can play a significant role, including being unemployed or having a low income because of the child’s health condition (83), ongoing adverse family relationship (83), and parental stress at followup (99).
These initial findings can provide an initial basis for interventions. Support needs to be aimed not only at the child but also at parents and caretakers. Quality of life evaluation should be incorporated systematically in outcome evaluations in people with congenital heart defects—ideally, longitudinally and prospectively. From a research perspective, there is an ongoing need for validating and improving quality of life tools targeted at people of appropriate age with congenital heart defects, and those culturally appropriate and also including the caretaker perspective. Incorporating quality of life in outcome assessment will also provide a more realistic assessment of benefits of treatment and prevention.
Outcomes: Gaps, Needs, Trends
As one examines and interprets the available data on outcomes, it is crucial to be mindful of their gaps and limitations. Missing or biased information is a poor and potentially damaging basis for action. Gaps throughout the lifespan of people with congenital heart defects, include even such crucial issues of social justice such as health disparities; these need to be actively investigated and resolved. In addressing data-related gaps and needs, it is helpful to consider trends—in pregnancy terminations, early detection, risk factor distribution—that will likely affect the health impact and care of congenital heart defects in the near future and would therefore benefit from ongoing monitoring.
Gaps Through the Lifespan
The gaps in our knowledge of outcome is particularly evident when taking a lifespan perspective of the experience and needs of people with congenital heart defects (Fig. 2.7).
Viewed in his light, it is easy to appreciate how issues wax and wane from conception (and indeed preconception) through all the stages of a person’s life. Initially, early diagnosis and optimal treatment are particularly crucial. As children grow into adults, developmental outcomes, employment, and social integration become increasingly important. From a primary prevention perspective, the crucial period is in very early pregnancy, and in fact, before conception. Other key aspects such as quality of life and costs are probably best evaluated over an entire lifespan, especially as life expectancy continues to improve.
The gaps are clear. Today’s data relate mainly to newborns and young children, and typically address outcome metrics in isolation (prevalence, mortality, costs). As a consequence, gaps are greater and data quality is poor as the focus shifts forward into adult life, or backward into pregnancy and preconception. These gaps have reasons. Birth and infancy experience the clinical urgency, personal drama, and substantial costs associated with diagnosis and early treatment. At the same time, data on these events are comparatively easy to collect—most encounters occur in a hospital or healthcare setting, followup is close, and databases are available. However, as one moves forward from infancy and childhood, measuring health outcomes become harder: not only the issues become more complex—involving quality of life, social integration, educational outcomes, and nonmedical costs—but typically they are captured incompletely, if at all, by commonly available data sources—for example, educational outcomes and quality of life are invisible to vital records, hospital discharge data, and most administrative data sets. As a consequence, the greatest information gaps are found precisely for those issues that are more complex and invest the greatest segment of the lifespan. At the same time, such an integrated, lifespan perspective is precisely what affected people, families, and health professional would need for comprehensive care and planning. Meeting this challenge is difficult. It will require a long term and coordinated investment in people, systems, and resources.
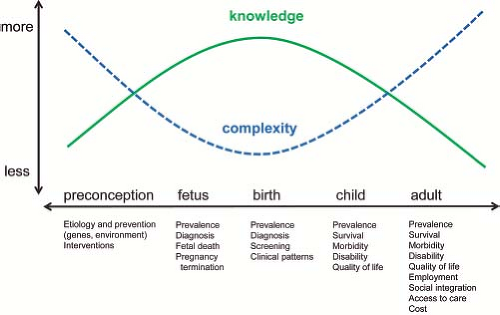 Figure 2.7 Lifespan perspective: Addressing the continuum of medical, social, and family issues from preconception to adulthood. |
Need for Accurate and Goal-Oriented Coding and Classification
Extracting information from data requires many steps. Data coding and classification is an early but critical phase. Typically, coding translates words from a clinical description into well-defined descriptive codes, which then can be used for data sharing and epidemiologic analysis. By moving from description to a limited set of codes, coding usually entails a loss of information. One challenge for a coding system is to retain the essential information while discarding the inessential. Clearly, errors, inconsistencies, and systematic variations will affect validity. The choice of the coding system is neither simple nor obvious. What is essential in one application may not be essential in others—information on surgical procedures or severity of a valvar stenosis is essential in an outcome study but less so in an etiologic study.
Compared to coding, classification requires special attention to the purpose of the study or survey. The goal is to generate meaningful analytic groups that can answer questions. The same (coded) cases can be classified differently depending on whether a study aims at evaluating the origin and causes of a heart defect (e.g., common pathogenesis linked to neural crest abnormalities), or monitoring health outcomes (e.g., mortality). Typically a specific instance of a heart defect may be assigned several multiple descriptive codes, whereas in many classification systems there is an emphasis to try and map each instance to one primary heart defect group. In practice, many coding systems are designed with a default classification structure, which is typically hierarchical and based on the code’s digits.
Coding and classification are crucial because they are the basis for all epidemiologic analyses: decisions, inconsistencies, or errors in coding and classification can affect findings significantly. In fact, part of the challenge of comparing and aggregating data is the variation or uncertainty in coding and classification among different studies.
As a practical illustration of real-life issues, it is helpful to briefly review two coding systems used in many applications—the World Health Organization (WHO)’s International Classification of Diseases (ICD) system, and the Society of International Surgeons (STS) system—and a classification system that focuses on pathogenesis, used in many etiologic studies (Table 2.1).
The WHO’s ICD system, in its different editions, is used internationally to generate epidemiologic data, including prevalence and mortality, for many diseases. The ICD system for congenital heart defects is mainly based on anatomy. The hierarchy of codes tends to group by regional anatomy. As an all-purpose system, with some emphasis on causes of mortality, the ICD system has known limitations in its ability to code the more complex cardiac defects or to distinguish degrees of severity of a given lesion. These limitations are particularly felt in studies of etiology and outcomes. The clinical modifications developed by the British Paediatric Association (BPA), now the Royal College of Paediatrics and Child Health (RCPCH), mitigate some of the limitations, and are used in many birth defect registries and in epidemiologic studies internationally.
TABLE 2.1 Coding and Classification Systems for Congenital Heart Defects: Examples | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
A welcome addition, particularly for studies of outcome, is the initiative by the International Society for Nomenclature of Paediatric and Congenital Heart Disease (100) that has led to the International Paediatric and Congenital Cardiac Code (101). Freely available (102), this coding system has features that allow capture of clinical correlates and procedures. Developed by a team of topic experts, this system has many strengths (Table 2.1), and is starting to be used in some birth defect surveillance and population surveys (103,104).
Other systems focus explicitly on classification, rather than coding. One of these systems (Table 2.1), developed by Dr. E. B. Clark (105,106), uses pathogenetic and mechanistic considerations (rather than anatomy or severity) to group most types of congenital heart defects in a few major analytic groups. The aim is to improve a study’s ability to identify associations between risk factors and congenital heart defects. In such studies, it is crucial to avoid defining groups so narrowly that each group has too few cases (thus losing statistical power and missing associations), while not incurring the opposite error, making groups so broad that heterogeneous conditions are lumped (thus also underestimating or missing associations). Because pathogenetic mechanisms (known or putative) are relatively few, sample size is increased while preserving the underlying homogeneity. In this system, each instance of congenital heart defects is assigned to one principal pathogenetic group (one baby, one classification), and these are ranked by presumed timing in development (Table 2.2). In its original form or with modifications, the pathogenetic classification has been used in several large epidemiologic studies, including the Baltimore-Washington Infant Study (BWIS) (15,16) and in some analysis of the Atlanta Birth Defects Case Control Study (107,108).
Assigning a single principal diagnosis is challenging. In the National Birth Defects Prevention Study (NBDPS), investigators added groups that described common associations (109). In an extension of the original system, the NBDPS classification system also stratifies phenotypes based on whether the heart defect is simple, an association, or complex (109). The purpose is to identify a subset of “pure” groups of heart defects, defined as simple phenotypes in individuals without extracardiac anomalies. By minimizing anatomic and pathogenetic heterogeneity, this approach aims at improving the ability to find meaningful associations with risk factors.
Need to Describe by Person, Place, and Time
The most useful information tends to be contextual—relevant to a specific population, place, and time. When trying to use findings from clinical or epidemiologic studies as a basis for recommendations and interventions, it is important to specifically assess not only the results but also the methods. For example, even a simple metric such as birth prevalence can be challenging (Table 2.3).
When evaluating prevalence it is very helpful to think in terms of person, place, and time. In practice, this means clearly defining first the “person,” namely, the study population and the heart defects (including method of coding and classification, inclusion, and exclusions) in addition to data sources and reporting procedures. Defining “place” is also important, because of well-known geographic variations in prevalence. One implication of these geographic variations is that when evaluating interventions it is crucial to have preintervention baselines that are local (this obvious step can be forgotten in the haste of implementing prevention strategies). Finally, the element of “time” is important not only as a reminder to assess temporal trends, but also to highlight the need to expand studies to adolescents, adults, and the elderly, so that their needs are better assessed and met.
Need to Identify and Resolve Health Disparities
Health disparities are a primary consideration in implementing and evaluating prevention activities (110). Disparities typically refer to differences in the occurrence, mortality, and burden of disease among groups of people. Disparities may arise because of the unequal distribution in the population of risk factors, including access to care, environmental exposures, low socioeconomic status, compounded or heightened by variations in disease susceptibility (110,111). Identifying and eradicating health disparities are critical steps in pursuing a measure of social justice in community health (110,111).
TABLE 2.2 Pathogenetic Classification of Heart Defects, with Proposed Hierarchy | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
One example of health disparity is the disproportionate mortality for congenital heart defects (and birth defects in general)
in developing countries compared to developed countries, as discussed above. However, health disparities also appear within developed countries, and are a major concern (110). For example, mortality for congenital heart defects in the United States, appears to vary by race and ethnicity; this has been documented for decades (50,52). Infant mortality in the United States, based on death certificate files, has been consistently higher for black infants than for white infants (52). For Hispanic infants, the pattern is less consistent, and rates are closer to those in white infants than reported previously (50,112).
in developing countries compared to developed countries, as discussed above. However, health disparities also appear within developed countries, and are a major concern (110). For example, mortality for congenital heart defects in the United States, appears to vary by race and ethnicity; this has been documented for decades (50,52). Infant mortality in the United States, based on death certificate files, has been consistently higher for black infants than for white infants (52). For Hispanic infants, the pattern is less consistent, and rates are closer to those in white infants than reported previously (50,112).
TABLE 2.3 Prevalence of Congenital Heart Defects: Person, Time, Place, and Implications for Prevention | ||||||||
|---|---|---|---|---|---|---|---|---|
|
These patterns can be complex (49), and mortality by race-ethnicity can vary by birthweight class. In one study, neonatal mortality attributable to heart defects was 20% higher among infants of black mothers compared with white mothers among term infants, but the reverse was true among preterm infants (49). Differences in outcomes by race and ethnicity have been found in followup studies after surgery (113) and in a population-based study of early childhood mortality (34)—in the latter, non-Hispanic black infants with severe heart defects were at a two-fold increased risk of dying compared to white infants.
Some of these differences in survival persist at all ages, others vary. In the United States, mortality in blacks compared to whites was disproportionately higher in infants and children (rate ratios of approximately 1.5 overall), but became similar or even lower in older adults (49), perhaps because of fewer survivors or fewer diagnoses at those ages. For Hispanics, the pattern is less clear, but following a similar pattern—increased mortality early in life compared to white infants, but similar or even lower mortality at older ages. Of note, increased mortality was reported in several US states (114) among children living in deprived communities, using community-level indicators of socioeconomic status.
The reasons for these variations are unclear and further studies are needed to understand, for example, whether they are due to reporting, prenatal diagnosis, prevalence of heart defects, or differences in risk factors for congenital heart defects or medical treatment. It is important to pursue these findings as they could indicate barriers to quality and timely care, potentially amenable to intervention and prevention.
This list of gaps and needs is necessarily selective. Many others could be discussed. A further element of complexity is the presence of trends and factors that will likely change the landscape of congenital heart defect, though at different rates across countries and healthcare systems. A few such trends are worth discussing, as they could have a significant impact on the lives of people and on communities.
Trends and Future
Congenital heart defects are common, costly, and critical. Predicting how this will change is difficult. It may be more helpful to briefly surmise some forces that will likely drive these changes, as these forces will vary in different places and times.
Changes in the total prevalence of congenital heart defects (the sum of live births, stillbirths, and pregnancy terminations) will depend on the balance of risk factors and protective factors in a population.
Some trends are not encouraging. Overall, the world is not getting healthier. Important risk factors for congenital heart defects are increasing. Diabetes, several chronic illnesses, and obesity are affecting more (and sometimes younger) people in many developed and developing countries. Also, demographic trends suggest an increasing maternal age at conception in many developed countries. This shift would lead to more pregnancies at higher risk of maternal-age dependent chromosomal syndromes (e.g., common trisomies), of which congenital heart defects are a common finding. The increase would likely be small on a yearly basis, and probably undetectable unless the data are examined carefully. The prevalence at birth of these heart defects would also depend on the concurrent use of fetal diagnosis and pregnancy termination. As a risk factor, the population distribution of maternal age is difficult to modify, though education and preconceptional counseling could have an impact in individual situations.
Yet, some factors offer hope: there appears to be now greater attention to integrated preconception health, and, in the areas of
chronic disease, as well as greater action in preventing, screening, or treatment of diabetes and obesity. Whether these initiatives will reverse the current worrying trends remains to be seen.
chronic disease, as well as greater action in preventing, screening, or treatment of diabetes and obesity. Whether these initiatives will reverse the current worrying trends remains to be seen.
Prenatal diagnosis will likely have a significant impact on occurrence and outcomes, though its direction and magnitude are not entirely predictable. In some areas, pregnancy terminations account for a substantial fraction of cases of selected congenital heart defects. In a study from Europe with data through 2005, 6% of all cases of heart defects not associated with chromosomal anomalies were terminations of pregnancy, with variations across registries and type of congenital heart defect (13). Notably, the reported rate of prenatal diagnosis was fairly low (13%) suggesting that as prenatal detection rates increase, terminations of pregnancy could account for a higher fraction of cases in the future. For some heart defects, the impact of pregnancy termination can be considerable. In the latest annual report (2012) from the International Clearinghouse for Birth Defects Surveillance and Research (Fig. 2.8), pregnancy terminations accounted in some countries for the majority of cases of hypoplastic left heart syndrome (29).
Clearly, failure to incorporate pregnancy terminations will underestimate the overall occurrence and impact of congenital heart defects. Failure to consider pregnancy terminations can also bias etiologic studies, for example, if an exposure (e.g., smoking or maternal illness) is associated with the likelihood of a pregnancy termination.
Diagnosis is typically easier than treatment; in countries with increasing medical technology, such as large parts of Asia and Africa, rates of prenatal diagnosis of severe heart defects will likely increase, at least initially without corresponding availability of effective and affordable treatment: depending on the social context, pregnancy terminations due to fetal anomalies could increase. Conversely, with better treatment options, family choices may change and birth prevalence of some severe heart defects could even increase, if cases previously terminated prenatally are allowed to reach birth. Prenatal diagnosis could also decrease morbidity and mortality if it decreases delayed diagnoses and promotes better organization of care.
Newborn screening for critical congenital heart defects using pulse oximetry is being implemented in several areas with the goal of detecting at birth certain critical congenital heart defects (115,116,117,118,119). Appropriately implemented, newborn screening should increase early diagnoses of some severe heart defects: the rationale is that outcomes should then improve, because treatment would start promptly (before closure of the ductus arteriosus) and in a well-prepared healthcare setting. From the perspective of outcome evaluation and birth prevalence, universal screening could promote the rapid and complete identification of the heart defects with greatest public health impact. While high-quality registries with multiple sources of ascertainment and extended followup would be unlikely to miss many such cases, the linkage between neonatal screening and epidemiologic surveillance could lead to significant improvements in data quality in the many areas with only basic registries or none at all. Appropriately leveraged, universal screening would create a valuable repository of information on selected major congenital heart defects for the entire screened population. Such population-based data would provide a powerful basis not only for monitoring prevalence, but also in assessing outcomes, conducting etiologic studies, and evaluating prevention interventions.
Trends in cost are difficult to predict. As more babies survive longer, their use of healthcare resources will likely increase. In general, congenital heart defects are likely to remain one of the more costly birth defects—by way of comparison, most children with orofacial clefts can be effectively “cured” by early surgery and lead an essentially normal and productive life, unlike many children with complex heart defects who will require long-term treatment and repeat surgeries. However, earlier diagnosis, through prenatal or newborn screening, could theoretically decrease costs if it substantially reduces preoperative morbidity and postoperative complications; to date, these benefits remain unproven.
Mortality associated with congenital heart defects is also likely to change. Deaths due to congenital heart defects (as for birth defects in general) will likely increase as a proportion of infant deaths, as infant deaths due to other causes (infections, prematurity) decline. In absolute terms, however, mortality should decrease gradually with better treatment and earlier diagnoses. Lower mortality will translate in greater longevity: prevalence in adults will increase, with greater needs for specialized care. Unless adequate services for older individuals are provided, the peak of mortality for heart defects risks being delayed rather than decreased. Finally, some factors may lead to an apparent change in mortality unrelated to true improvements in outcomes. For example, increased pregnancy terminations for fetal cardiac defects can cause an apparent reduction in mortality as a proportion of the population (as it is usually tracked using death certificate data), because fewer babies will be born with heart defects and would be at risk for dying. Including pregnancy terminations in birth defect surveillance would help avoid this bias. Conversely, as screening and diagnostic technology is introduced in a country in which it had not been available previously, deaths
attributed to heart defects (and attributed to other causes previously) may increase simply because of better ascertainment.
attributed to heart defects (and attributed to other causes previously) may increase simply because of better ascertainment.
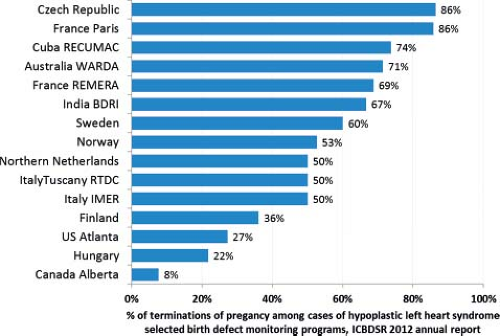 Figure 2.8 Proportion of elective terminations of pregnancy among all cases of hypoplastic left heart syndrome reported in selected birth defect monitoring programs, International Clearinghouse for Birth Defects Surveillance and Research, 2012. (Data from International Clearinghouse for Birth Defects Surveillance and Research. Annual Report 2012. In: Mastroiacovo P, ed. Rome Italy; 2012.) |
Trends and Future: Implications for the Detection of the Next Epidemic
Most of the factors discussed so far (e.g., newborn screening, pregnancy terminations) would lead to trends whose detection and explanation would be relatively straightforward. A different concern is the unexpected, unpredictable introduction in a population or region of a teratogen—for example, retinoic acid—causing a cluster of congenital heart defects. Early detection of such teratogen-induced “epidemics” of birth defects is a stated goal of many monitoring programs. Medications and environmental exposures are particular concerns for the general public and the ability to respond to these concerns quickly and in a cost-efficient manner is a significant benefit of having a high-quality monitoring system in place.
Effective monitoring must balance the ability to detect true changes (high sensitivity, low false-negative rates) with the cost of investigating false alarms (false-positives). This requires a system that is able to select, among the continuous stream of monitoring signals, those with the greatest epidemiologic and biologic plausibility. Epidemics can be missed by setting the bar too high (the signal is not picked up) or too low (because limited resources are spread across too many futile investigations). Practical challenges include the presence of local or global trends that can shift background rates, missing cases by not ascertaining pregnancy terminations, and low-quality data when diagnosis is based on administrative data sets (e.g., vital records only). Rising to these challenges requires increased resources and innovative approaches, some of which are summarized in Table 2.4.
These approaches strive to improve the quality of clinical description and cardiology expertise available to monitoring programs, and to implement a structured, accurate, and rapid response to a concerning “signal.” Universal neonatal screening, if linked to public health monitoring, could rapidly expand population-based ascertainment of most severe congenital heart defects, thus facilitating the surveillance for clusters. In addition, pediatric cardiologists can play an important role as the “astute clinicians,” who note unusual occurrences (e.g., rare defects clustering in time and space) and activate the public health system for further triaging. This function is particularly helpful in the presence of small clusters, which are otherwise difficult to detect promptly, if at all. The subsequent epidemiologic investigations could identify new or emerging causes of congenital heart defects and prevent further epidemics.
Evidence for Action: Epidemiology of Risk and Causes
Effective primary prevention starts with characterizing modifiable causes of congenital heart defects in human populations: Characterizing causes means that causality is satisfactorily established, and the associated risk is qualified and quantified. Some risk factors, such as diabetes or retinoic acid, reach this threshold of evidence and are prime candidates for prevention. Others are less well characterized: causality may be in question (e.g., obesity) or the magnitude of risk may be unclear (e.g., lithium), typically because of either insufficient data or inconsistent findings leading to concerns for bias and confounding.
A major challenge in characterizing risk factors in human populations (as opposed to experimental models) is the near-exclusive reliance on observational studies—randomized clinical trials are just not possible for presumed noxious exposures or maternal illnesses. Exceptions may include putative protective factors such as folic acid supplementation, for which risk reduction is expected and no material side effects are anticipated. Findings in observational studies—typically case-control studies, rarely cohort studies—can be distorted by bias, confounding, or statistical noise, resulting in data that can be confusing and difficult to interpret. For this reason, it is helpful to first review some key epidemiologic concepts in risk
assessment, as a general framework to help navigate the complex body of etiologic studies relative to congenital heart defects (1,120).
assessment, as a general framework to help navigate the complex body of etiologic studies relative to congenital heart defects (1,120).
TABLE 2.4 Detecting the Next Epidemic of Heart Defects: Tools and Value | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
What Does it Mean to Characterize Modifiable Causes?
To form the basis for interventions, the evidence on modifiable causes needs to be robust and accurate. In practice, it is helpful to systematically address some key questions (Table 2.5).
The first key question—causality—is perhaps the most challenging. Observational studies typically generate associations. Moving from associations to causality is a significant step that requires thoughtful evaluation—causality is a complex concept (121)—supported by evidence that goes well beyond the results of a single study, however well-conducted. Particularly for observational studies, support for causality can be strengthened by finding consistent, plausible associations from multiple well-designed studies that stringently control for confounding and convincingly minimize bias. Confounding and bias are significant concerns. An association between maternal smoking and congenital heart defects could be due to confounding by alcohol use, if alcohol causes congenital heart defects and is more common among smokers compared to nonsmokers. Such association with smoking could also be due purely to bias: For example, recall bias can occur in a case-control study if mothers of affected babies are more likely than mothers of controls to remember or report smoking during pregnancy. Biases may not only create but also hide association: This can occur in particular when exposures are misclassified (nondifferentially), as can occur when based solely on maternal reports without validation or use of biomarkers. Finally, in addition to confounding and bias, associations may also be due to chance. This is more of a concern in smaller studies, in which random variations due to sampling can occur more easily. Though chance effects are of concern, they are fairly easy to measure and manage, whereas confounding and bias can be difficult to prevent, detect, and eliminate. Finally, other forms of bias can influence what is reported in the literature: Publication bias, for example, may favor the emergence of “positive” versus “null” findings and needs to be explicitly considered when reviewing and summarizing the body of published evidence.
Specificity is an important but often underappreciated element of evidence. Specific associations can provide clues to pathogenesis and strengthen the case for causality. The relatively specific association of retinoic acid exposure to complex conotruncal defects not only helped identify it as a cardiac teratogen but pointed toward a developmental effect on neural crest cells. However, specificity is not and need not be always present: established teratogens such as maternal diabetes can cause a variety of heart defects. Identifying specific associations requires careful study design in order to collect an unbiased sample of specific phenotypes, and the input of expert clinicians in case of review and classification. Finally, information on specificity is useful when assessing the potential benefits for prevention—a risk factor for hypoplastic left heart syndrome would be viewed differently from one causing a small atrial septal defect.
Magnitude of risk can be estimated in case-control and cohort studies, in the form of relative risk of disease, that is, the ratio of disease risk in the exposed divided by the risk in the unexposed. Some relative risks may be low (e.g., smoking), others can be considerable (maternal diabetes), and a few can be extremely elevated (e.g., retinoic acid). Ideally, one would also estimate the absolute risk for congenital heart defects, that is, the absolute rate of disease in those exposed (the numerator of relative risk). Absolute risk is important information in individual counseling. For example, the relative risk for heterotaxy could be 10 (i.e., risk among the exposed is 10 times the risk among the unexposed); however, if the baseline risk in the unexposed is low, say 1 in 10,000, then the absolute risk,
though increased, is still in the order of 1 in 1,000 (or 999 to 1 odds of not developing that heart defect).
though increased, is still in the order of 1 in 1,000 (or 999 to 1 odds of not developing that heart defect).
TABLE 2.5 Characterizing Modifiable Risk Factors for Congenital Heart Defects | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


