Chapter 26 Epidemiology and Pathophysiology of Mesenteric Vascular Disease
Severe acute intestinal ischemia results from sudden symptomatic reduction in intestinal blood flow of sufficient magnitude to potentially result in intestinal infarction.1 Acute ischemia of the small bowel and/or right colon may result from mesenteric arterial occlusion (embolus or thrombosis), mesenteric venous occlusion, and nonocclusive processes, especially vasospasm (Fig. 26-1). Isolated dissections of the superior mesenteric artery (SMA), either in association with cystic medial degeneration of the arteries or, more commonly, as progression of an existing dissection in the descending thoracic aorta into the SMA and celiac artery, may also result in acute intestinal ischemia.
Acute Arterial Occlusive Mesenteric Ischemia
Acute Mesenteric Arterial Embolism
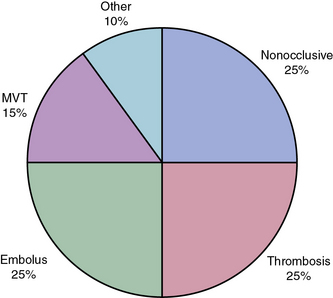
Roughly 25% of all cases of acute mesenteric ischemia are due to emboli to the SMA, 25% of cases are due to thrombosis of preexisting atherosclerotic lesions, and the remaining 50% are due to a variety of other etiologies.2 Mesenteric emboli can originate from left atrial or ventricular mural thrombi or from cardiac valvular lesions.1 These thrombi are most often associated with cardiac dysrhythmias such as atrial fibrillation, global myocardial dysfunction with poor ejection fraction, or discrete hypokinetic regions produced by previous myocardial infarction (MI).3 About 15% of emboli lodge at the origin of the SMA, but the majority lodge 3 to 10 cm distally in the tapered segment of the SMA just past the origin of the middle colic artery.2 More than 20% of emboli to the SMA are associated with concurrent emboli to another arterial bed.4 Intestinal ischemia due to embolic arterial occlusion can be compounded by reactive mesenteric vasospasm, further reducing collateral flow and exacerbating the ischemic insult.2,5
Acute Mesenteric Arterial Thrombosis
Thrombosis of the SMA or the celiac artery is usually associated with preexisting critical stenoses. Many of these patients have histories consistent with chronic mesenteric ischemia (CMI), including postprandial pain, weight loss, “food fear,” and early satiety.1,6 Superior mesenteric artery thrombosis can be regarded as a complication of untreated chronic intestinal ischemia.1 The SMA plaque likely progresses slowly to a critical stenosis over years until thrombosis occurs.
Unlike embolic occlusions, thrombosis of the SMA generally occurs flush with the aortic origin of the vessel. Aortic dissection involving the visceral vessels, though rare, can cause acute mesenteric ischemia. The intimal flap of the dissection can extend into, compress, or exclude the mesenteric orifice.5 Acute mesenteric ischemia is also an uncommon (< 1%) but serious complication of cardiac surgery, with a reported mortality rate of greater than 50% in most series.5,7 Presumably, the nonpulsatile perfusion delivered by most extracorporeal circuits allows severely stenotic visceral vessels to occlude during cardiopulmonary bypass. Identified risk factors for this complication include prolonged cross-clamp times, use of intraaortic balloon counterpulsation, low cardiac output syndromes, blood transfusion, triple-vessel disease, coronary artery disease (CAD), and peripheral artery disease (PAD).7
Pathophysiology of Occlusive Acute Mesenteric Ischemia
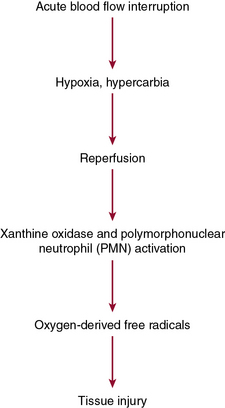
Acute mesenteric ischemia, whether the underlying cause is embolic or thrombotic, may eventually lead to intestinal infarction (Fig. 26-2). Hypoxia and hypercarbia that occur during flow interruption, and reperfusion injury once intestinal blood flow is restored, all contribute to tissue loss8 (Fig. 26-3). Reperfusion injury is believed to be principally mediated by activation of the enzyme xanthine oxidase and recruitment and activation of circulating neutrophils (PMNs).9 The mechanism of injury likely involves production of oxygen-derived free radicals by xanthine oxidase that then causes profound local tissue injury through lipid peroxidation, membrane disruption, and increased microvascular permeability.5,9 The ischemic endothelium recruits PMNs in an autocrine and paracrine manner by secreting chemotactic cytokines (tumor necrosis factor [TNF]-α, interleukin [IL]-1, platelet-derived growth factor [PDGF]) that perpetuate further damage to the reperfused tissue. Once activated, PMNs degranulate, releasing myeloperoxidase, collagenases, and elastases that further injure already ischemic and vulnerable tissue.5,10 Activation of this endogenous inflammatory cascade is not restricted to the injured organ and may also have deleterious systemic effects, with cardiac, pulmonary, and other organ system dysfunction.5
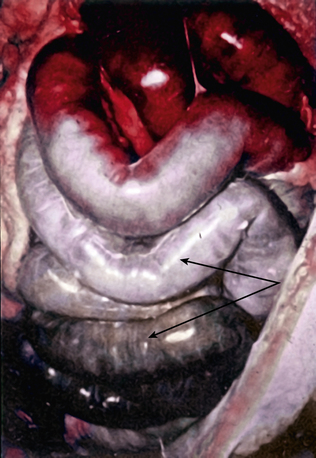
Natural History of Acute Mesenteric Arterial Occlusive Disease
The mortality rate for occlusive acute mesenteric ischemia exceeds 70% in most series.1,2,5,11,12 Occlusive acute intestinal ischemia resulting from SMA embolism has a more favorable prognosis than that resulting from SMA thrombosis.1 Survival following acute intestinal ischemia due to SMA thrombosis is rare. The more favorable prognosis associated with embolism is attributable to the fact that most emboli lodge distally in the SMA (beyond the origin of the middle colic artery), thus allowing perfusion of the proximal intestine via middle colic and jejunal artery branches.1 Thrombotic occlusion of the SMA usually occurs proximal to the middle colic artery, and therefore completely interrupts mid-gut arterial perfusion in patients with poorly developed celiac artery or inferior mesenteric artery (IMA) collateral flow.1,3,6
Nonocclusive Mesenteric Ischemia
Epidemiology
Nonocclusive mesenteric ischemia (NOMI) accounts for 25% of all episodes of acute intestinal ischemia.1,5,13,14 In NOMI, microscopic arterial blood flow is inadequate to supply perfusion to the bowel. The result is intestinal ischemia and infarction in the presence of a patent macroscopic vasculature.15 Previous reports15–17 have identified multiple risk factors for development of NOMI (Box 26-1). Mesenteric arterial vasospasm may occur following elective revascularization procedures for chronic SMA occlusion. In such cases, vasoconstriction of small and medium-sized vessels is precipitated by early enteral feeding.18 Without prompt intervention, NOMI may progress from localized intestinal ischemia to transmural infarction, peritonitis, and death.19,20 Mortality is high regardless of treatment, owing to the underlying medical conditions that precipitate NOMI and frequent delays in diagnosis.5,13–15
 Box 26-1
Box 26-1Pathophysiology
Nonocclusive mesenteric ischemia was first recognized in autopsies of patients with small-intestinal gangrene in the absence of arterial or venous occlusion.21,22 Investigation of the regulatory mechanisms of mesenteric circulation has demonstrated that the pathophysiology of NOMI is multifactorial. Virtually all patients with NOMI have a severe coexisting illness, commonly severe cardiac failure.23 It is postulated that hypoperfusion from cardiac failure, resulting in peripheral hypoxemia and splanchnic vasoconstriction, precipitates intestinal ischemia.13,14 Mesenteric vasoconstriction, intestinal hypoxia, and ischemia-reperfusion injury all contribute to development of NOMI.
Mesenteric vasoconstriction, the hallmark of NOMI, represents an exaggerated homeostatic mechanism induced by excessive sympathetic activity during cardiogenic shock or hypovolemia. The body attempts to maintain cardiac and cerebral perfusion at the expense of splanchnic and peripheral circulations. Experimental evidence suggests that the mediators of this response are endothelin-1 (ET-1), nitric oxide (NO), vasopressin, and angiotensin (Ang).24,25 Endothelin-1 is a potent vasoconstrictor secreted from endothelial cells (ECs); in concert with other vasoactive peptides, it regulates myogenic cells in the vascular wall. Nitric oxide can have paradoxical effects on vascular tone, depending on local concentration.26 At low concentrations it acts as a vasodilator, whereas at higher concentrations it acts as an oxygen-derived free radical, impairing mitochondrial energy production.
The splanchnic autoregulatory system is affected by local arteriolar smooth muscle relaxation and vasodilation, as well as increased cellular oxygen extraction.27 Adequate oxygen delivery may be maintained despite declining perfusion pressures until a critical threshold is reached. In experimental models, maximal extraction is reached at a pressure of 40 mmHg, but beyond this point, oxygen consumption declines, and ischemia ensues.28 Neri et al. recently demonstrated that postoperative cardiac surgery patients who develop NOMI had persistent deficits between oxygen delivery (DO2) and consumption (VO2) because of poor circulatory reserve. In contrast, postoperative cardiac patients who did not develop NOMI were able to normalize their DO2:VO2 ratio by optimizing their cardiac output.14 In the presence of impaired perfusion, blood flow is not evenly distributed in the bowel wall. The mucosa retains its perfusion at the expense of the serosal layers through mucosal production of NO, prostaglandins (PGs), and stimulation of dopamine-I receptors.27 Histological damage is first observed at the villous tip and progresses to the deeper muscularis, submucosa, and mucosa within a few hours.
Once set in motion, mesenteric vasospasm may persist despite correction of the precipitating event.13,14 The etiology of persistent vasoconstriction once adequate blood flow is restored is unknown, but it may respond to direct intraarterial papaverine infusion or other vasodilators, including iloprost.29 This phenomenon of protracted vasoconstriction, however, plays an important role in development and maintenance of occlusive and nonocclusive intestinal ischemia, and may also complicate mesenteric revascularization.18
Use of vasoconstrictor agents and digitalis has been associated with the majority of cases of NOMI. Vasoactive agents, including α-adrenergic drugs and vasopressin, produce splanchnic vasoconstriction directly, whereas digoxin preparations alter mesenteric vasoreactivity by stimulating arterial and venous smooth muscle cell (SMC) contraction.27,29 This may enhance mesenteric arteriolar vasoconstriction in the setting of acute venous hypertension.29
Restoration of blood flow to the ischemic intestine may be complicated by reperfusion injury. During critical ischemia, adenosine triphosphate (ATP) levels are depleted, causing distortion of ATP-dependent cell membrane systems. This results in loss of cellular homeostasis, with cellular swelling and electrolyte imbalances.27 Reduction in ATP levels also generates large amounts of adenosine, a precursor of hypoxanthine. Within the swollen cells, calcium accumulates and triggers hydrolysis of the enzyme xanthine dehydrogenase into xanthine oxidase, which reacts with intracellular hypoxanthine to produce uric acid and toxic oxygen free radicals.5,27 These free radicals exert direct damage to cellular membranes, causing capillary leak syndrome, and incite endogenous inflammatory cascades that cause widespread tissue injury. The deleterious effects of free radicals are usually limited by endogenous scavengers such as glutathione, catalase, superoxide dismutase, and NO.27 However, in cases of prolonged ischemia, the capacity of this scavenger system to eliminate reactive oxygen species (ROS) is exceeded, and continued damage occurs. The degree of reperfusion injury is thus related to the frequency and duration of the ischemic episodes. Clark and Gewertz demonstrated that two short 15-minute periods of low flow followed by reperfusion resulted in a more severe histological injury than a single 30-minute period of ischemia.30,31
In NOMI, a similar scenario exists: hypoperfusion may be partial and occasionally repetitive. It is believed that episodic reperfusion creates a local environment replete with primed neutrophils within the ischemic bed that are capable of degranulating and releasing superoxide. This concept is substantiated by recent experimental evidence that reperfusion injury may be attenuated by reperfusion with leukodepleted blood or by blockade of EC surface receptors for leukocyte adherence.32 In addition, several compounds including N-acetylcysteine and vitamin E have been shown in animal models to reduce tissue damage caused by reactive oxygen species.33 Application of these novel approaches in human NOMI awaits further study.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


