Eosinophilic lung disease of undetermined cause
Idiopathic eosinophilic pneumonias
Idiopathic chronic eosinophilic pneumonia
Idiopathic acute eosinophilic pneumonia
Eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome)
Hypereosinophilic syndrome
Idiopathic hypereosinophilic obliterative bronchiolitis
Eosinophilic lung diseases of determined cause
Eosinophilic pneumonias of parasitic origin
Tropical eosinophilia
Ascaris pneumonia
Eosinophilic pneumonia in the larva migrans syndrome
Strongyloides stercoralis infection
Eosinophilic pneumonias in other parasitic infections
Eosinophilic pneumonias of other infectious causes
Allergic bronchopulmonary aspergillosis and related syndromes
Allergic bronchopulmonary aspergillosis
Other allergic bronchopulmonary syndromes associated with fungi or yeasts
Bronchocentric granulomatosis
Drug, toxic agents, and radiation-induced eosinophilic pneumonias
Drugs (typical, occasional, or exceptional eosinophilic pneumonia)
Toxic agents (toxic oil syndrome, L-tryptophan)
Eosinophilic pneumonia induced by radiation therapy to the breast
Miscellaneous lung diseases with possible associated eosinophilia
Organizing pneumonia
Asthma and eosinophilic bronchitis
Idiopathic interstitial pneumonias
Pulmonary Langerhans cell histiocytosis
Malignancies
Other
Eosinophil Biology
Initially thought to be especially important in the defence against parasitic infestation, eosinophil leukocytes are now considered multifunctional cells implicated in innate and adaptive immunity, including but not restricted to numerous inflammatory reactions to parasitic helminth, bacterial, and viral infections [1]. Their broad role in homeostasis function, physiology, and pathophysiology is now well appreciated.
Eosinophil precursors differentiate and mature in the bone marrow under the action of cytokines and especially of interleukin (IL)-5, IL-3, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [1, 2]. Activation of the Δdbl-GATA-1 transcription factor is deemed critical in this process. Mature eosinophils then circulate in the blood for about 1 day before being attracted into tissues, successively involving chemotaxis, adhesion, and diapedesis and processes, under the control of IL-5 and eotaxin-1. In the tissues, they undergo apoptosis unless survival factors (mostly IL-5) are present.
The eosinophil contains two types of intracytoplasmic granules, the content of which can be released by degranulation, while other mediators are secreted (with involvement of vesicle-associated membrane proteins in the regulation of granule fusion within the cell). The larger granules, identified by a dense crystalloid matrix at electron microscopy, contain the characteristic cationic proteins major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil-derived neurotoxin (EDN), and the enzymatic protein eosinophil peroxidase (EPO) [1, 2]. The smaller amorphous granules contain arylsulfatase and acid phosphatase. The process of degranulation by activated eosinophil releases cationic proteins into the extracellular space, with potential direct toxicity to the heart, brain, and bronchial epithelium. Degranulated eosinophils can be identified at electron microscopy by presence of cytoplasmic vacuoles, and loss of electron density of the central core of the granules (inversion or disappearance of core density). Molecular and intracellular pathways regulating eosinophil differentiation, priming, activation, degranulation, and mediator secretion, and how the release of toxic substances contributes to the pathophysiology of eosinophilic disorders, have become better understood on a molecular standpoint and are reviewed elsewhere [2]. In addition to cationic proteins, eosinophils release proinflammatory cytokines, lipid- and arachidonic acid–derived mediators, enzymes, reactive oxygen species, and matrix metalloproteases, all of which may contribute to the pathophysiology of eosinophilic lung diseases.
The eosinophil is involved in many allergic or inflammatory processes through its interaction with other cells, including especially T helper (Th) lymphocytes, but also mast cells and basophils, endothelial cells, macrophages, platelets, and fibroblasts. Intercellular signaling is mediated by surface expression of adhesion molecules, apoptotic signaling molecules, chemokines, complement receptors, chemotactic factor receptors, cytokine receptors, and immunoglobulin receptors. For instance, eosinophils are capable of regulating mast cell function and histamine release. Eosinophils have further immune properties. They express the major histocompatibility complex II protein human leukocyte antigen (HLA)–DR, can present the antigen to T-helper lymphocytes, and secrete an array of cytokines, thereby promoting effector T-cell proliferation. They can synthetise IL-4 and promote IL-4, IL-5, and IL-13 secretion by CD4+ T cells (promoting Th2 lymphocyte activation), and secrete indoleamine 2,3-dioxygenase (indirectly promoting Th1 apoptosis), modulating the Th1/Th2 balance. Abnormalities in the T-cell receptor repertoire and T-cell clonotype of BAL lymphocytes and peripheral blood lymphocytes seem to contribute to the pathophysiology of eosinophilic lung diseases [3]. Overall, the paradigm of eosinophil function has changed from a terminal effector cell in allergic airway diseases to their being involved in the initial stages of pathophysiology.
Corticosteroids shorten eosinophil survival in the blood and tissues, and are currently the most potent drugs to treat eosinophilic disorders, although lack of specificity can result in numerous adverse events. As the recruitment of eosinophils to the lung mostly implicates IL-5 and the eotaxin subfamily of chemokines (itself regulated by the Th2 cell–derived IL-13 cytokine), those are the target of several drugs in development, which can potentially change dramatically the therapeutic landscape of eosinophilic disorders in the near future [4]. Other promising targets for therapy include the IL-5 receptor, CD2 binding protein, IgE, and IL-4/IL-13 receptor, with a few agents already at the stage of clinical evaluation. Although histopathologic lesions in eosinophilic pneumonias are largely reversible, tissue damage can occur especially in allergic bronchopulmonary aspergillosis (ABPA), and in eosinophilic granulomatosis with polyangiitis (EGPA) (Churg-Strauss syndrome, CSS), with possible remodeling and fibrosis in the bronchial mucosa. It is unknown yet whether anti-IL-5 antibodies and other therapeutic agents that target eosinophil-specific molecules, may prevent tissue damage. In addition, newer drugs specifically targeting cytokines of the eosinophil lineage contribute to a better understanding of the pathogenic role of eosinophils [4].
General Features of Eosinophilic Pneumonias
Historical Perspective
Early descriptions of pulmonary “infiltration with eosinophilia” [5], of “pulmonary eosinophilia” [6], and later of “cryptogenic pulmonary eosinophilias” [7] included cases now considered as probable idiopathic chronic eosinophilic pneumonia (ICEP), EGPA, and Löffler’s syndrome. Carrington and colleagues [8] described in 1969 the syndrome of ICEP.
Clinical Presentation
Eosinophilic pneumonia is a pneumonia where the eosinophils are the most prominent inflammatory cells on histopathologic examination, whereas infiltration with lymphocytes and neutrophils is moderate. Eosinophilic pneumonias are separated into two main etiologic categories: (1) those with a definite cause; and (2) idiopathic eosinophilic pneumonias, either solitary or associated with extrathoracic manifestations that are the hallmark of EGPA, but can also be observed, to a lesser extent, in hypereosinophilic syndromes (HES), drug reactions, or infections especially parasitic infections. It is therefore mandatory that the clinician take a full history and search thoroughly for a cause with potentially practical consequences, such as parasitic infection or drug or toxic exposure.
Most eosinophilic pneumonias can be classified within one of the well-characterized and individualized syndromes. They may manifest by different clinicoradiologic syndromes, namely Löffler’s syndrome, chronic eosinophilic pneumonia, or acute eosinophilic pneumonia, mostly differing from one another by the pattern of disease onset, severity, and evolution with or without corticosteroid treatment. The vast majority of cases of eosinophilic pneumonia respond dramatically to corticosteroid treatment and heal without significant sequelae.
Pathology
Open lung biopsies that used to be performed for the diagnosis of ICEP have provided material for the few histopathologic studies published on eosinophilic pneumonia [7–9]. The pathologic features described in ICEP represent a common denominator of all categories of eosinophilic pneumonias, whatever their origin. Additional specific features may be observed depending on the etiological context (e.g. bronchocentric distribution of lesions in ABPA; presence of parasites or fungal hyphae in eosinophilic pneumonia of parasitic origin).
In ICEP, the alveolar spaces are filled with eosinophils representing the predominant inflammatory cell, together with a proteinaceous and fibrinous exudate, respecting the global architecture of the lung. The distribution of eosinophilic pneumonia is generally diffuse. Macrophages are also present in the infiltrate, with scattered multinucleated giant cells occasionally containing eosinophilic granules or Charcot-Leyden crystals [8]. An associated interstitial inflammatory cellular infiltrate is invariably present, consisting of eosinophils, lymphocytes, plasma cells, and histiocytes. Some eosinophilic microabscesses may be observed (foci of necrotic intra-alveolar eosinophils surrounded by macrophages or epithelioid cells with palisading arrangement). Degranulated eosinophils can be identified within the site of eosinophilic pneumonia by electron microscopic or immunohistochemical studies [10]. Areas of non-prominent organization of the alveolar inflammatory exudate are common [8]. Mucus plugs obstructing the small airways may be present in ICEP [8] and especially in ABPA. A mild non-necrotizing vasculitis involving both small arteries and venules is common, however necrosis and fibrosis are absent.
In idiopathic acute eosinophilic pneumonia (IAEP), the pathologic pattern includes intra-alveolar and interstitial eosinophilic infiltrates, diffuse alveolar damage, intra-alveolar fibrinous exudates, organizing pneumonia, and non-necrotizing vasculitis [11].
Diagnosis
The clinical diagnosis of eosinophilic pneumonia is suspected in patients with respiratory symptoms (dyspnea, cough, or wheezing), pulmonary opacities at chest imaging, and eosinophilia demonstrated in the peripheral blood or (preferably) in the lung.
Surgical or video-assisted thoracoscopic lung biopsy is seldom necessary. Although they can show characteristic features of eosinophilic pneumonia, transbronchial lung biopsies are generally not recommended due to the small size of the specimen that allows only partial morphologic evaluation. Bronchoalveolar lavage (BAL) is now considered a good surrogate of lung biopsy to demonstrate lung eosinophilia, although no study has definitely established a correlation between increased eosinophils at differential cell count and eosinophilic pneumonia at lung pathology. In normal subjects, BAL eosinophilia is lower than 1 % of cells at differential count. In contrast, BAL eosinophilia greater than 40 % is found mainly in patients with chronic eosinophilic pneumonia, whereas BAL eosinophilia between 3 and 40 % (and especially between 3 and 9 %) may be found in various interstitial lung diseases other than eosinophilic pneumonia. A conservative cutoff of 40 % of eosinophils at BAL differential cell count has been adopted for the diagnosis of ICEP in clinical studies [12, 13], and a cutoff of 25 % has been proposed for the diagnosis of IAEP [14]. We recommend that a clinical diagnosis of eosinophilic pneumonia be supported by alveolar eosinophilia when the eosinophils (1) are the predominant cell population of BAL cell count (macrophages excepted) and (2) represent more than 25 % of differential cell count (with greater specificity when greater than 40 %).
Blood eosinophilia or hypereosinophilia when present also contributes to the diagnosis of eosinophilic pneumonia in a patient with compatible HRCT features. It may be missing in patients who have already received systemic corticosteroids, and it is often absent at presentation in IAEP. Blood cell count must thus be done before starting corticosteroids. Blood eosinophilia is defined by an eosinophil blood count greater than 0.5 × 109/L, and hypereosinophilia by an eosinophil blood count greater than 1.5 × 109/L on two examinations at least 1 month interval, and/or tissue hypereosinophilia [15]. Blood eosinophilia greater than 1 × 109/L (and preferably hypereosinophilia) may obviate the need to perform BAL in individual cases with typical presentation. For example, BAL may occasionally be omitted to confirm Löffler’s syndrome (as it occurs in ascariasis) in a patient with mild cough, wheezes, transient pulmonary opacities at chest radiograph, and frank blood eosinophilia. However BAL is generally useful to rule out alternative diagnoses (such as bacterial or parasitic pneumonia, or pulmonary infiltrates related to Hodgkin disease), and BAL is recommended to confirm the diagnosis of eosinophilic pneumonia in most cases.
Eosinophilic Lung Disease of Undetermined Cause
ICEP is characterized by a progressive onset of symptoms over a few weeks with cough, increasing dyspnea, malaise, and weight loss, whereas IAEP presents as an acute pneumonia (similar to acute lung injury or acute respiratory distress syndrome [ARDS]) with frequent respiratory failure necessitating mechanical ventilation. Both conditions are idiopathic.
Idiopathic Chronic Eosinophilic Pneumonia
Chronic eosinophilic pneumonia was first described in detail by Carrington and colleagues [8], in a series of nine patients, and was further confirmed and detailed by several and numerous case reports.
Clinical Features
ICEP predominantes in women with a 2:1 female-to-male ratio [9, 13], with a peak of incidence in the fourth decade [9], and a mean age of 45 years at diagnosis [13]. A majority of patients with ICEP are nonsmokers [9, 13], suggesting that smoking might be protective. About half of the patients have a history of atopy [9, 13] and up to two thirds have a history of asthma [9, 12, 13, 16, 17], with no particularities in the clinical presentation of ICEP with the exception of higher total immunoglobulin (Ig) E levels in asthmatics [12]. In addition, asthma may develop concomitantly with the diagnosis of ICEP (15 % of patients) or develop after ICEP (about 15 % of patients) [12]. Asthma in patients with ICEP often gets worse and requires long-term oral corticosteroid treatment [12].
ICEP is characterized by the progressive onset of cough, dyspnea, and chest pain [9, 13], with a mean interval between the onset of symptoms and the diagnosis of 4 months [13]. Mechanical ventilation may be required on exceptional occasion. Hemoptysis is rare but can occur in up to 10 % of cases [9, 13]. Chronic rhinitis or sinusitis symptoms are present in about 20 % of patients [13]. At lung auscultation, wheezes are found in one third of patients [9] and crackles in 38 % [13]. Systemic symptoms and signs are often prominent, with fever, weight loss (>10 kg in about 10 %), and commonly asthenia, malaise, fatigue, anorexia, weakness, and night sweats.
Imaging
The imaging features of ICEP are characteristic, although they may overlap with those found in cryptogenic organizing pneumonia. Peripheral opacities at chest x-ray present in almost all cases [8, 9, 13, 18, 19] consist of alveolar opacities with ill-defined margins, with a density varying from ground-glass to consolidation (Fig. 15.1), and are migratory in 25 % of patients [13]. The classic pattern of “photographic negative or reversal of the shadows usually seen in pulmonary edema,” highly evocative of ICEP, is seen in only one fourth of patients [9], however peripheral and upper zone predominance of abnormalities is usually present.
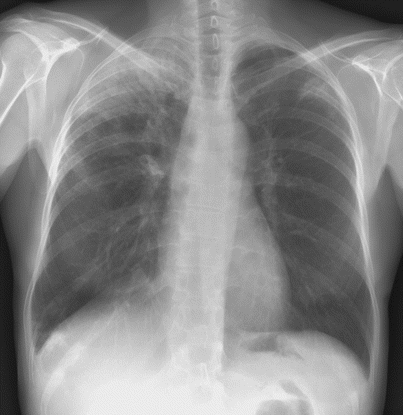
Fig. 15.1
Chest radiograph of a patient with idiopathic chronic eosinophilic pneumonia showing peripheral alveolar opacities predominating in the right upper lobe
Whereas the opacities are bilateral in at least 50 % of cases at chest x-ray [9], the proportion of bilateral opacities increases up to more than 95 % at high-resolution computed tomography (HRCT) [13] (Fig. 15.2). Predominance of ground-glass attenuation and consolidation in the periphery and upper lobes of the both lungs [9, 13] is very suggestive of ICEP [13, 19, 20]. Septal line thickening is common [20]. Centrilobular nodules (less than 20 % of cases) [19], consolidation with segmental or lobar atelectasis, can also be seen. Upon corticosteroid treatment, consolidation rapidly decreases in extent and density, possibly evolving to ground-glass attenuation or inhomogeneous opacities, and later to streaky or bandlike opacities parallel to the chest wall. Cavitary lesions are extremely rare and should lead to reconsideration of the diagnosis. Reverse halo sign that is very suggestive of organizing pneumonia is rare contrary in ICEP. Pleural effusions (which are common in IAEP) are rare and usually mild or moderate in ICEP. Mediastinal lymph node enlargement may be seen in 15–20 % of cases [13].
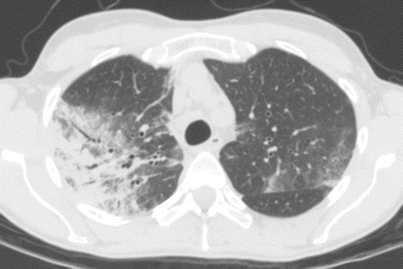
Fig. 15.2
Computed tomography (CT) scan of a patient with idiopathic chronic eosinophilic pneumonia showing bilateral asymmetric peripheral alveolar opacities with airspace consolidation and ground glass opacity
Laboratory Studies
Peripheral blood eosinophilia is a diagnostic criterion of ICEP, and therefore the proportion of patients with ICEP and possible normal peripheral blood count is unknown. The mean blood eosinophilia was 5.5 × 109/L in our series [13]. Eosinophils represent 26–32 % of the total blood leukocyte count [9, 13]. C-reactive protein level is elevated [9, 13]. Total blood IgE level is increased in about half of cases and greater than 1,000 kU/L in 15 % [13]. Antinuclear antibodies may occasionally be present [13]. Urinary EDN level indicating active eosinophil degranulation is markedly increased [21].
Bronchoalveolar Lavage
BAL eosinophilia is constant and key to the diagnosis of ICEP, obviating the need for lung biopsy in the vast majority of cases (Table 15.2). The mean eosinophil percentage at BAL differential cell count was 58 % at diagnosis in the series from our group [13], however the eosinophil count drops within a few days upon corticosteroid treatment. The percentage of neutrophils, mast cells, and lymphocytes a BAL may also be increased [13]. Sputum eosinophilia may also be present.
Table 15.2
Diagnostic criteria for idiopathic chronic eosinophilic pneumonia
1. Diffuse pulmonary alveolar consolidation with air bronchogram and/or ground glass opacities at chest imaging, especially with peripheral predominance; |
2. Eosinophilia at BAL differential cell count ≥40 % (or peripheral blood eosinophilia ≥1.0 × 109 /L>; |
3. Respiratory symptoms present for at least 2–4 weeks; |
4. Absence of other known causes of eosinophilic lung disease (especially exposure to a drug susceptible to induce pulmonary eosinophilia) |
BAL eosinophils of patients with ICEP show features of cell activation and release eosinophil proteins, which are phagocytosed by macrophages. ECP and EDN levels are increased in the BAL fluid. Eosinophils are recruited to the lung through various chemokines, and are resistant to Fas-induced apoptosis. Eosinophilic activation may be compartmentalized to the lung, as expressed by differential expression of HLA-DR molecules between alveolar and blood eosinophils. BAL lymphocytes include CD4+ memory T-cells (expressing CD45RO+, CD45RA−, CD62L−), and may present clonal rearrangement of the T-cell receptor repertoire [3].
Differential Diagnosis
Extrapulmonary manifestations when present should challenge the diagnosis of ICEP and especially to consider EGPA or overlap between ICEP and EGPA. Arthralgias, repolarization (ST-T) abnormalities on the electrocardiogram, pericarditis, altered liver biologic tests, eosinophilic lesions at liver biopsy, mononeuritis multiplex, diarrhea, skin nodules, immune complex vasculitis in the skin, and eosinophilic enteritis have been occasionally reported in ICEP [8, 13]. Furthermore, eosinophilic pneumonia may be a presenting feature of EGPA; corticosteroid treatment prescribed for ICEP may prevent the subsequent development of overt systemic vasculitis.
Lung Function Tests
An obstructive ventilatory defect is present in about half the patients [9, 13], and a restrictive ventilatory defect in the other half [13]. The CO transfer factor is decreased in half of patients, and the transfer coefficient in about one fourth. Hypoxemia (PaO2 <75 mmHg) present in two thirds of patients [13] may be due to right-to-left shunting in consolidated areas of the lung, as suggested by increased alveolar-arterial oxygen gradient [9]. With treatment, the lung function tests rapidly return to normal in most patients [9]. However, a ventilatory obstructive defect may develop over years in some patients, especially those with a markedly increased BAL eosinophilia at initial evaluation [22].
Treatment and Prognosis
Because most patients receive corticosteroids, the natural course of untreated ICEP is not well known [9]. However, spontaneous resolution of ICEP may occur [9, 13]. The clinical and radiologic response to corticosteroids is dramatic, with improvement of symptoms within 1 or 2 weeks and even within 48 h in about 80 % [13] of cases, and rapid clearance of pulmonary opacities at chest x-ray. In one series, the chest radiograph was significantly improved at 1 week in 70 % of patients, and almost all had a normal chest x-ray at their last follow-up visit [13]. Death directly resulting from ICEP is exceedingly rare.
The optimal dose of corticosteroids is not established, but treatment may be initiated with 0.5 mg/kg/day of prednisone, with slow tapering over 6–12 months based on clinical evaluation and blood eosinophil cell count. Most patients require treatment for longer than 6–12 months because of relapse in more than half of patients while decreasing below a daily dose of 10–15 mg/day of prednisone, or after stopping oral corticosteroids treatment [9, 13]. Relapses respond very well to corticosteroid treatment, that usually can be resumed at a dose of about 20 mg/day of prednisone [13].
The clinical series in which long-term follow-up is available clearly show that most patients need very prolonged corticosteroid treatment: in a series with a mean follow-up of 6.2 years, only 31 % were weaned at the last control visit [13]. Relapses of ICEP must be distinguished from asthma symptoms, and are less frequent in asthmatics, possibly because of inhaled corticosteroids prescribed after stopping oral corticosteroids [12, 13]. Inhaled corticosteroids might thus help in reducing the maintenance dose of oral corticosteroids, although they are not effective enough when given as monotherapy [23]. Long-term steroid use may lead to osteoporosis. Omalizumab, a recombinant humanized monoclonal antibody against IgE, was reported to prevent recurrence of ICEP and to spare oral corticosteroids in case reports, however caution must be exerted given recent reports of omalizumab-associated EGPA [24, 25]. The anti-IL-5 monoclonal antibody mepolizumab has not yet been evaluated in patients with ICEP.
Idiopathic Acute Eosinophilic Pneumonia
IAEP is often misdiagnosed as infectious pneumonia because of fever and bilateral opacities on chest x-ray present in all patients. However, IAEP [11, 14, 17, 26–29] markedly differs from ICEP by its acute onset, the severity of hypoxemia, the usual lack of increased blood eosinophils at presentation contrasting with highly increased eosinophil percentage at BAL, and the absence of relapse after clinical recovery. As AEP can also be due to drug exposure or infection, known causes of acute eosinophilic lung disease must be excluded for the diagnosis of IAEP to be made (Table 15.3).
Table 15.3
Diagnostic criteria for idiopathic acute eosinophilic pneumonia
1. Acute onset of febrile respiratory manifestations (≤1 month duration before consultation) |
2. Bilateral diffuse opacities on chest radiography |
3. Hypoxemia, with PaO2 on room air <60 mmHg, and/or PaO2/FiO2 ≤300 mmHg, and/or oxygen saturation on room air <90 % |
4. Lung eosinophilia, with >25 % eosinophils on BAL differential cell count (or eosinophilic pneumonia at lung biopsy) |
5. Absence of infection, or of other known causes of eosinophilic lung disease (especially exposure to a drug susceptible to induce pulmonary eosinophilia) |
Clinical Features
IAEP may present at any age [30], however the mean age at presentation is around 30 years [14, 30], with a very strong predominance in males [29]. Most patients have no prior asthma history [17]. However, taking a thorough exposure history is mandatory, as a causative role of cigarette smoke is established. Most patients have been recently exposed to dust or cigarette smoke within the days before onset of disease, and often will have begun to smoke, restarted to smoke, or increased the number of cigarettes smoked daily, especially within 1 month before the onset of “idiopathic” AEP [29, 31]. The disease is therefore often not “idiopathic”, being initiated or triggered by inhaled nonspecific causative agents in susceptible individuals, however it can occur in the absence of any inhaled exogenous trigger. AEP may develop soon after the initiation of smoking especially when starting with large quantities, and may relapse – not always – in patients who resume cigarette smoking [29, 31]. Flavoring components of smoked cigars have been suspected. In addition, the onset of IAEP seem to follow in some patients outdoor activities or peculiar exposures, such as cave exploration, plant repotting, wood pile moving, smokehouse cleaning, motocross racing in dusty conditions, indoor renovation work, gasoline tank cleaning, explosion of a tear gas bomb, or exposure to World Trade Center dust [14, 30, 32].
IAEP develops acutely or subacutely over less than 1 month in previously healthy individuals, with cough, dyspnea, fever, and chest pain at presentation [11, 30]. More than half of patients present with acute respiratory failure [29]. Abdominal complaints and also myalgias can occur [14]. Clinical signs include crackles or, less often, wheezes, and tachypnea and tachycardia.
Imaging
Imaging of patients with IAEP is quite distinct from those with ICEP. In addition to bilateral alveolar and/or interstitial opacities (Fig. 15.3) [14, 27, 28, 30], the chest x-ray commonly shows bilateral pleural effusion and Kerley B lines [14]. The chest x-ray returns to normal within 3 weeks [14, 30], with pleural effusions being the last abnormality to disappear [14]. Typical computed tomography (CT) abnormalities include ground-glass attenuation and air space consolidation (Fig. 15.4), with poorly defined nodules. Interlobular septal thickening and bilateral pleural effusion seen in a majority of patients are highly suggestive of the diagnosis in the setting of eosinophilic pneumonia [14, 18, 27, 30, 33] (or in a patient spuriously suspected to have infectious pneumonia).
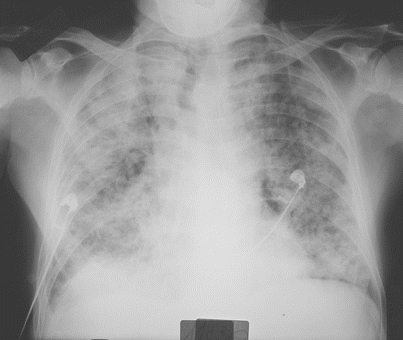
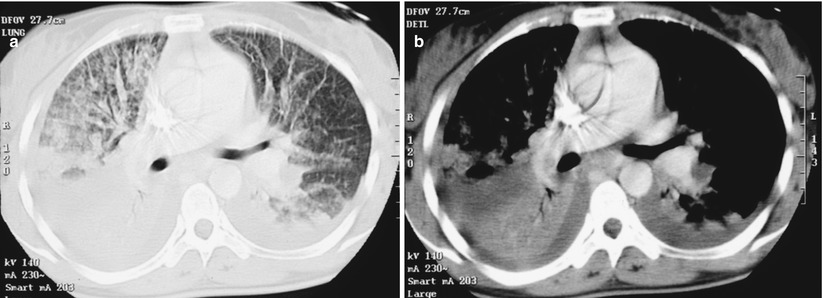
Fig. 15.3
Chest radiograph of a patient with idiopathic acute eosinophilic pneumonia and acute respiratory failure showing diffuse alveolar consolidation
Fig. 15.4
CT scan of a patient with idiopathic acute eosinophilic pneumonia showing bilateral diffuse alveolar consolidation with air bronchogram and ground-glass opacity (a, parenchymal window) and bilateral mild pleural effusion (b, mediastinal window)
Laboratory Studies
In contrast with ICEP, peripheral blood eosinophilia is usually lacking at presentation, with white blood cell count showing increased leukocyte count with a predominance of neutrophils. However, the eosinophil count often rises within days during the course of disease [14, 17, 30], a retrospective finding very suggestive of IAEP. Eosinophilia is also present at pleural fluid differential cell count [14] and in the sputum [17]. The IgE level may be elevated. Serum levels of thymus and activation-regulated chemokine (TARC/CCL17) (and other biomarkers including KL6 and exhaled nitric oxide) are often increased in IAEP, however lack of specificity of this finding does not rule out acute lung injury or infectious pneumonia [34, 35].
Bronchoalveolar Lavage
BAL is the key to the diagnosis of IAEP, especially in patients without blood eosinophilia at presentation. The finding of greater than 25 % eosinophils at BAL generally obviates the lung biopsy, at least in immunocompetent patients The average percentage of eosinophils at BAL differential count varies between series (37 % [14] to 54 % [30]), and lymphocyte and neutrophil counts can be moderately increased. Importantly, systematic bacterial cultures of BAL fluid are sterile, and appropriate stainings are negative, ruling out infectious agents that can cause AEP. After recovery, eosinophilia at BAL may persist for several weeks.
Lung Function Tests
Hypoxemia may be severe in patients with IAEP, a majority of whom fit the definition of ARDS of various severity (except that there is no known clinical insult identified in IAEP), e.g. acute onset of respiratory failure not fully explained by cardiac failure or fluid overload (with objective exclusion of hydrostatic edema), bilateral opacities (not fully explained by effusions, lobar/lung collapse, or nodules), and a PaO2/FiO2 [fractional inspired oxygen concentration] <300 mmHg with positive end-expiratory pressure or continuous positive expiratory pressure ≥5 cm H2O [36]. However, shock is exceptional and extrapulmonary organ failure does not occur in IAEP, in sharp contrast with IAEP.
Hypoxemia is associated with right-to-left shunting in areas with consolidation, and may be refractory to breathing 100 % oxygen in some patients [26, 30]. Alveolar-arterial oxygen gradient is increased [14]. Although mechanical ventilation was necessary in a majority of patients in earlier series [14, 30], more recent series have shown that the severity of IAEP is more varied than originally reported [29].
When performed in less severe cases, lung function tests show a mild restrictive ventilatory defect with normal forced expiratory volume in 1 s–to–forced vital capacity (FEV1/FVC) ratio and reduced transfer factor. After recovery, lung function tests are generally normal, with possible ventilatory restriction in some of them [14].
Lung Biopsy
Lung biopsy is seldom necessary when BAL demonstrates alveolar eosinophilia. In older series of patients with IAEP, lung biopsy has shown acute and organizing diffuse alveolar damage together with interstitial alveolar and bronchiolar infiltration by eosinophils, intra-alveolar eosinophils, and interstitial edema [11, 14, 37].
Treatment and Prognosis
Exclusion of possible causes of AEP, especially infections and drugs, is key to the management of patients with AEP. Recovery of IAEP can occur without corticosteroid treatment [30, 34], and therefore improvement concomitant with corticosteroid treatment is not a diagnostic criterion of IAEP. In most patients diagnosed with IAEP, a corticosteroid treatment is initiated, with initially intravenous methyl prednisolone later changed to oral prednisone or prednisolone that can be tapered over 2–4 weeks [14]. FiO2 may be decreased within a few hours of corticosteroid treatment in many patients initially requiring oxygen [14]; most patients are rapidly weaned from the ventilator. The clinical improvement begins within 3 days [29]. The chest x-ray is normalized within 1 week in 85 % of patients, but mild pulmonary infiltrates and pleural effusion may still be present at CT at 2 weeks [29]. One recent study of 137 patients suggested that a treatment duration of 2 weeks may be sufficient, with an initial daily dose of 30 mg of prednisone (or 60 mg of intravenous methylprednisolone every 6 h in patients with respiratory failure) [29]. No relapse occurs after stopping corticosteroid treatment, in contrast with ICEP (Table 15.4).
Table 15.4
Distinctive features between idiopathic chronic eosinophilic pneumonia (ICEP) and idiopathic acute eosinophilic pneumonia (IAEP)
ICEP | IAEP | |
|---|---|---|
Onset | >2–4 weeks | <1 month |
History of asthma | Yes | No |
Smoking history | 10 % of smokers | 2/3 of smokers, often recent initiation |
Respiratory failure | No | Usual |
Initial blood eosinophilia | Yes, on admission | No (delayed) |
BAL eosinophilia | >25 % (generally >40 %) | >25 % |
Chest imaging | Homogeneous peripheral airspace consolidation | Bilateral patchy areas of ground glass attenuation, airspace consolidation, interlobular septal thickening, bilateral pleural effusion |
Predominance in upper lobes and lung periphery | ||
Relapse | Yes, possibly multiple | No |
No significant clinical or imaging sequelae persist on the longer term. Mortality is rare despite the frequent initial presentation with acute respiratory failure. Identification of causative tobacco or environmental exposures is key to preventing rare recurrences, that in most cases are due to resuming of cigarette smoking after smoking cessation.
Eosinophilic Granulomatosis with Polyangiitis (Ex Churg-Strauss Syndrome)
History and Nomenclature
The first reliable case of EGPA was reported by Lamb in 1914 [38]. Churg and Strauss described in 1951 [39] the eponymous syndrome of “allergic granulomatosis, allergic angitiis, and periarteritis nodosa”, mainly from autopsied cases. In the 1992 Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis [40], CSS was included in the group of small vessel vasculitides. The nomenclature of the systemic vasculitides was revised in 2012 at the international Chapel Hill consensus conference [41], and the terminology of CSS was replaced by EGPA. As antineutrophil cytoplasmic antibodies (ANCA) are present in about 40 % of the cases, EGPA belongs to the pulmonary ANCA-associated vasculitides, together with microspic polyangiitis and granulomatosis with polyangiitis (Wegener’s), and together with single organ ANCA-associated vasculitis.
EGPA is defined as an eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium vessels, and associated with asthma and eosinophilia [41]. The disease may be confined to a limited number of organs especially the upper or lower respiratory tract [41]. The terminology of EGPA underscores that it is indeed a vasculitis, although not all patients have robust criteria of documented systemic vasculitis or ANCA [42]. The current terminology and classification likely requires further refinement.
Pathology
The pathologic lesions of EGPA (CSS) observed in recent series [43, 44] only rarely comprise all the characteristic features on biopsies from organs other than the lung, which is now rarely biopsied. The diagnosis is made earlier in the course of disease, often before overt vasculitis has developed, characterized histopathologically by eosinophilic infiltration of the tissues and often perivascular eosinophils but without vasculitis. In cases with overt EGPA, typical histopathologic features include vasculitis (necrotizing or not, involving mainly the medium-sized pulmonary arteries), granulomatous eosinophilic infiltration, and extravascular granuloma with palisading histiocytes and giant cells. When present, the eosinophilic pneumonia in EGPA is similar to ICEP.
Clinical Features
EGPA is a very rare systemic disease, with no sex predominance, predominating in adults younger than 65 [45, 46], with cases occasionally reported in children and adolescents. Asthma occurs at a mean age of about 35 years [45], preceding the onset of vasculitis by 3–9 years [45, 47–49]; therefore the mean age at diagnosis of EGPA ranges from 38 to 49 years [45, 49]. The interval between asthma and the onset of vasculitis may be much longer in rare cases [47], or they may be contemporaneous [49]. Asthma is generally severe, and frequently requires oral corticosteroids; its severity typically increases progressively until the vasculitis develops, but it may attenuate when the vasculitis flourishes (possibly as a result of corticosteroids) and further increase once the vasculitis recedes [45, 47].
Chronic rhinitis (75 % of cases) [45], relapsing paranasal sinusitis (60 %) [48], and nasal polyposis with eosinophilic infiltration at histopathology are frequent. Crusty rhinitis may be present, however it is much less severe in EGPA than in granulomatosis with polyangiitis. Septal nasal perforation and saddle nose deformation are exceedingly rare.
Asthenia, weight loss, fever, arthralgias, and myalgias often herald the onset of the systemic vasculitis.
Heart damage in EGPA is undoubtedly a major source of morbidity and mortality, although its onset is often insidious and asymptomatic and diagnosed only when left ventricular failure and dilated cardiomyopathy have developed, possibly leading to cardiac failure or sudden death [45–50]. Heart involvement mostly results from eosinophilic myocarditis, and rarely from arteritis of the larger coronary arteries [51, 52]. Although marked improvement usually occurs with corticosteroid treatment, heart involvement in EGPA may require heart transplantation, with possible recurrence of eosinophilic vasculitis in the transplanted heart. A strict cardiac evaluation is therefore warranted in any patient with suspected EGPA, generally including electrocardiogram, echocardiography, serum level of troponin, and magnetic resonance imaging of the heart. Cardiac MRI frequently shows late enhancement of the myocardium [53–55], which may correspond to myocarditis, however the absence of a gold standard, it is difficult at a given point in time to differentiate irreversible scar lesions from active inflammation requiring intense immunosuppression, and incidental findings from clinically relevant myocardial involvement. Treatment decisions are eventually based on critical clinical evaluation, taking into account results from several investigations, including electrocardiogram, echocardiography, troponine level, and possibly a combination of cardiac MRI and positron emission tomography. In addition to myocardial involvement, asymptomatic pericarditis with limited effusion at echocardiography is common, with rare cases of tamponade, and the risk of venous thromboembolic events [56] is increased in patients with EGPA. Endomyocardial involvement (typically seen in idiopathic HES) is uncommon in EGPA.
Mononeuritis multiplex, present in 77 % of patients [48], is the most frequent and the most typical of peripheral neurologic involvement in EGPA, which may also consist of asymmetrical polyneuropathy in the lower extremities, or rarely cranial nerve palsies or central nervous system involvement. Digestive tract involvement (31 % of cases [48]) consists in isolated abdominal pain, and less frequently intestinal or biliary tract vasculitis, diarrhea, ulcerative colitis, gastroduodenal ulcerations, perforations (esophageal, gastric, intestinal), digestive hemorrhage, or cholecystitis. Cutaneous lesions (50 % of patients[48]) mainly consist of palpable purpura of the extremities (Fig. 15.5), subcutaneous nodules (especially of the scalp and extremities), erythematous rashes, and urticaria. Renal involvement (about 25 % of cases) consists in mild severity glomerulonephritis or glomerular hematuria [48], however renal failure is rare contrasting with the other ANCA-associated vasculitides.
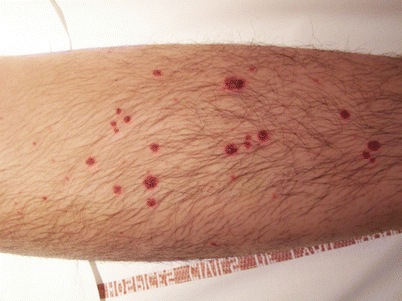
Fig. 15.5
Palpable purpura of the forearm in a patient with eosinophilic granulomatosis with polyangiitis
Imaging
Pulmonary opacities corresponding to eosinophilic pneumonia are present on chest x-ray in a majority of patients with EGPA (37 % [48] to 72 % [45, 57]) and consist of ill-defined opacities, sometimes migratory, transient, and of varying density [45, 47, 58, 59]. In contrast to GPA, pulmonary cavitary lesions are exceptional. The chest x-ray may remain normal throughout the course of the disease. Mild pleural effusion and phrenic nerve palsy can be observed. On thin-section CT, ground-glass attenuation and air space consolidation predominate, with peripheral or random distribution (Fig. 15.6). Bronchial wall thickening or dilation, interlobular septal thickening, hilar or mediastinal lymphadenopathy, pleural effusion, or pericardial effusion [18, 58, 59] are less commonly found. In one study, centrilobular nodules were more frequent in EGPA than in patients with ICEP [19]. However, EGPA is difficult to differentiate from other causes of eosinophilic lung diseases on the basis of HRCT imaging [18]. Importantly, pleural effusion when present may correspond to either inflammatory eosinophilic exudate directly related to EGPA or to a transudate caused by cardiomyopathy.
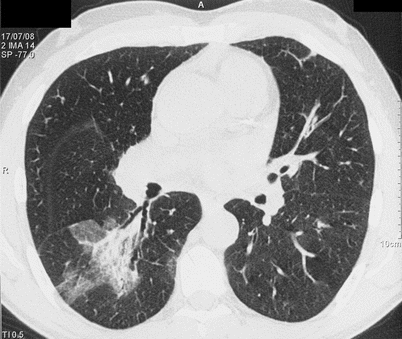
Fig. 15.6
CT scan of a patient with eosinophilic granulomatosis with polyangiitis showing airspace consolidation and ground-glass opacity in the right lower lobe
Laboratory Studies
Peripheral blood eosinophilia is a major feature of EGPA, with generally eosinophil counts comprised between 5 and 20 × 109/L and occasionally higher values [45, 47, 48]. Blood eosinophilia usually parallels disease activity, and disappears within hours after the initiation of corticosteroid treatment. Eosinophilia, sometimes greater than 60 %, is also found on BAL differential cell count and in the pleural fluid when present.
Although EGPA belongs to the group of ANCA-associated vasculitides, ANCAs are present in only about 40 % of patients. ANCAs in EGPA are mainly perinuclear (p-ANCA) with myeloperoxidase specificity, and rarely cytoplasmic ANCAs (c-ANCA) with proteinase 3 specificity [48, 49, 60–62]. Although nonspecific and not validated as useful biomarkers, the serum IgE level, the erythrocyte sedimentation rate, and the C-reactive protein level, and serum levels of IgG4, CCL17/TARC, and CCL26/Eotaxin-3 are increased. Anemia is common. High levels of urinary EDN may represent an activity index of disease.
Pathogenesis
EGPA is considered an autoimmune process involving T cells, endothelial cells, and eosinophils. Defects have been identified in regulatory CD4+ CD25+ or CD4+ CD25− T-cell lymphocytes (producing IL-10 and IL-2) that may influence progression of disease, and supporting an immunological hypothesis of disease. Furthermore, clonal CD8+/Vβ+ T cell expansions with effector memory phenotype and expressing markers of cytotoxic activity were found in peripheral blood lymphocytes, as well as T cell receptor-beta gene rearrangement. Patients carrying the major histopathology complex DRB4 allele, involved in acquired specific immunity, are prone to develop EGPA. Familial EGPA has been reported.
Contrasting to common belief, evidence of allergy demonstrated by specific IgE together with a corresponding clinical history is present in less than one third of patients. When present in EGPA, allergy mainly consists of perennial allergies to Dermatophagoides, whereas seasonal allergies are less frequent than in the general asthmatic patient [63].
Some vaccines or desensitization may trigger or play a role as adjuvant factors [64]. Other possible triggering factors include Aspergillus, allergic bronchopulmonary candidiasis, Ascaris, bird exposure, or smoked cocaine. Some drugs have been suspected to induce EGPA, especially sulfonamides (used together with antiserum), diflunisal, macrolides, diphenylhydantoin, and more recently the anti-IgE antibody omalizumab [65]. In addition, leukotriene-receptor antagonists (montelukast, zafirlukast, pranlukast) have been suspected to be involved in the development of EGPA, although their role is controversial [49, 66–70]. The occurrence of EGPA is more frequent in patients receiving leukotriene receptor antagonists, however there is conflicting evidence whether the association is coincidental, whether some cases of smoldering EGPA flare because of reducing oral or inhaled corticosteroids, or whether these drugs really exert a direct facilitating or triggering role on the vasculitis [24, 68]. A possible mechanistic link has been proposed [71]. Since EGPA may follow montelukast treatment in asthmatic patients without smoldering EGPA, may recur on rechallenge with the drug, and may remit on its withdrawal [66, 68], a causal relationship cannot be excluded [70], and we advocate that leukotriene receptor antagonists be avoided in patients with asthma, eosinophilia, and/or established or smoldering extrapulmonary manifestations.
Diagnosis
The classical description of EGPA follows three stages: asthma and rhinitis; tissue eosinophilia (such as a pulmonary disease resembling ICEP); and extrapulmonary eosinophilic disease with vasculitis. Diagnosing EGPA may be challenging in patients with early disease corresponding to the so-called formes frustes [72], who often already receive oral corticosteroids for asthma, thereby masking the underlying smoldering vasculitis. The diagnosis is more straightforward at a later stage of disease with overt systemic manifestations, however it is extremely important that the diagnosis be established before severe organ involvement (especially cardiac) is present.
There are currently no established diagnostic criteria for EGPA. Lanham and associates [45] have proposed three diagnostic criteria including (1) asthma, (2) eosinophilia exceeding 1.5 × 109/L, and (3) systemic vasculitis of two or more extrapulmonary organs, however those do not include ANCAs, which when present do contribute to the diagnosis. Classification criteria (which are not diagnostic criteria) have been proposed by the American College of Rheumatology [73] (Table 15.5), but they cannot be readily used for diagnosis in an individual with suspected EGPA. Provisional diagnostic criteria including ANCA have been recently proposed [42]. Although a pathologic diagnosis is desirable and can be obtained from the skin, nerve, or muscle [48], it is not mandatory in patients with characteristic features of EGPA. Because cutaneous lesions are easy to access (when not involving the face), a skin biopsy is commonly performed to obtain pathologic evidence of vasculitis when they are present (See Clinical Vignette). Conversely, lung biopsy either transbronchial or video-assisted is seldom done.
Table 15.5
Diagnostic and classification criteria of eosinophilic granulomatosis with polyangiitis
Lanham and colleagues [45] |
Asthma |
Eosinophilia |
Evidence of vasculitis involving at least two organs |
American College of Rheumatology [73] |
Asthma |
Eosinophilia >10 % |
Mononeuropathy, or polyneuropathy |
Pulmonary infiltrates, nonfixed |
Paranasal sinus abnormality |
Extravascular eosinophil infiltration on biopsy findings |
NB diagnosis is probable when four of the six criteria are present (sensitivity of 85 %, specificity of 99.7 %); these are classification criteria that may be used when the diagnosis of systemic vasculitis has been established by histopathology |
1992 Chapel Hill Consensus conference definition [40]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|