(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
ATP supplies in endothelial cells are relatively independent of mitochondrial oxidative pathways. Endothelial mitochondria emerge as agents with roles in modulating the dynamics of intracellular calcium and the generation of reactive oxygen species (ROS) and nitric oxide (NO). Oxidative stress and ischemia provoke an opening of the mitochondrial permeability transition pore and activation of mitochondrial pathways of apoptosis which compromises survival of endothelial cells. Impairment of mitochondrial function contributes to the development of endothelial dysfunction leading eventually to several diseases, such as atherosclerosis, diabetes, heart failure, and reperfusion injury associated with ischemia.
Introduction
Endothelial cells (ECs) represent the “frontline” cells against vascular disease. An essential “plumbing” role of the vasculature in facilitating the delivery of oxygen and energetic substrates for powering the high metabolic demands of the cardiomyocytes is obvious. The second vital role of the endothelium is a functioning as an impermeable barrier between the blood and the cardiac tissue. Damage of endothelium can lead to leukocyte and platelet extravasation, inflammation, vascular damage, and atherosclerosis. Damage of the vasculature during the development of the several types of cardiovascular disease highlights critical functions of the endothelium. A third important function of the vascular endothelium is the trophic support of cardiomyocytes and regulation of vascular tone by releasing of paracrine-signaling peptides (angiopoietin-1, neuregulin, platelet-derived growth factor, vascular endothelial growth factor) as well as signaling molecules (reactive oxygen species [ROS], nitric oxide [NO], adenosine).
In this chapter, we discuss the mechanisms by which mitochondria contribute to endothelial physiology and pathophysiology, focusing especially on following aspects:
Role of OXPHOS and mitochondrial Ca2+ in the release of vasodilators
The generation of ROS and NO
EC as targets of oxidative damage in the vasculature and their role in activation of the mitochondrial cell death pathways, such as apoptosis and necrosis
Release of Vasodilators: Role of OXPHOS and [Ca2+]m
One of the major functions of endothelium is the regulation of blood vessel tone via release of vasodilatory factors (NO, prostacyclin). ECs secrete vasodilators spontaneously and in response to several agonists—acetylcholine, adenine nucleotides (ATP, ADP), thrombin, histamine, and bradykinin [1]. Long-term secretion of vasodilators and a large production of L-arginine for NO synthesis require sustained supply of energy in the form of ATP. Accordingly, EC demands for ATP will increase in cases of agonist-accelerated secretion of relaxing factors (Fig. 7.1).
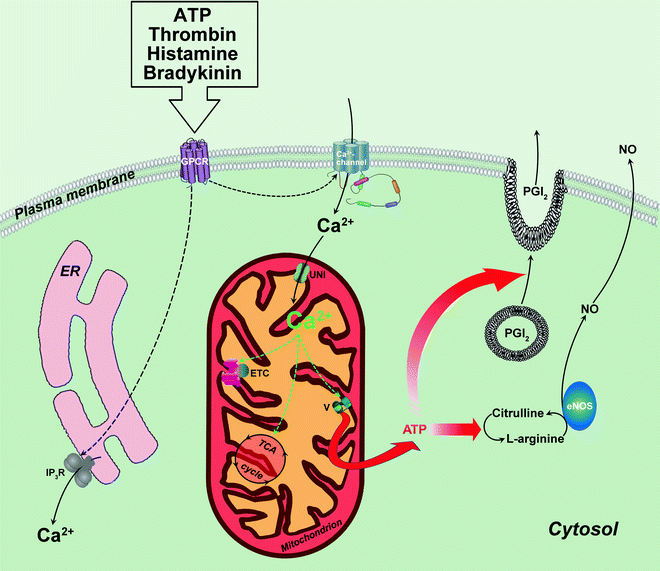
Fig. 7.1
ATP production in endothelial mitochondria on demand. Role of mitochondrial Ca2+. Agonists-accelerated production/secretion of relaxing factors in endothelial cell is accompanied by the elevation of cytoplasmic and mitochondrial Ca2+ concentration. Mitochondrial Ca2+ increase depends on extracellular Ca2+ influx through Ca2+ channel. Within mitochondrion, Ca2+ activates several key Ca2+-sensitive enzymes that participate in oxidative ATP synthesis (depicted by green dashed arrows). By this way, mitochondrial Ca2+ regulates mitochondrial ATP production to compensate for ATP utilization by endothelial cells actively producing nitric oxide (NO) and secreting vasodilators [prostacyclin (PGI2) is shown]. Other abbreviations: eNOS endothelial nitric oxide synthase; ER endoplasmic reticulum; ETC electron transport chain; GPCR G protein-coupled receptor; IP 3 R inositol 1,4,5-trisphosphate receptor; TCA tricarboxylic acid; UNI mitochondrial calcium uniporter; V ATP synthase. Translocations of molecules and metabolic conversions are shown by solid arrows; regulatory effects are shown by dashed arrows
Production of relaxant factors in endothelium appears to be dependent on mitochondrial OXPHOS [2], and changes in cytosolic and mitochondrial Ca2+ provide an efficient mechanism for regulation of ATP synthesis in accordance with EC demands. It is well documented that all extracellular substances regulating vasodilatory activity of ECs also elevate endothelial cytoplasmic Ca2+ concentration [3]. Typically they raise intracellular calcium about ten times within seconds, and then calcium concentration rapidly declines to a lower value. The initial fast agonist-stimulated rise of calcium concentration results from IP3-dependent mobilization of it from endoplasmic reticulum (ER), whereas slower sustained phase of calcium elevation is abolished by removal of extracellular calcium, suggesting that this phase results from calcium influx [3].
∼25% of intracellular Ca2+ is sequestered in the mitochondria of EC [4], and mitochondrial Ca2+ ([Ca2+]m) rises in association with agonist-evoked intracellular Ca2+ [5, 6]. Thus, increase of intracellular Ca2+ can be transferred to the mitochondrial matrix leading to the activation of several key Ca2+-sensitive mitochondrial enzymes that participate in oxidative ATP synthesis (Ca2+-sensitive dehydrogenases, ATP synthase—see “Chap. 2: mito & other organnels” for details). By this way, [Ca2+]m regulates mitochondrial ATP production to compensate for increased ATP utilization by actively secreting EC (Fig. 7.1).
Lawrie et al. [5] added important details to the mechanism of agonist-dependent regulation of endothelial [Ca2+]m. By using ATP as an agonist mobilizing intracellular Ca2+ via purinergic P2Y receptor, they demonstrated that in EC [Ca2+]m increase largely depends on extracellular Ca2+ influx, whereas agonist-induced IP3-dependent mobilization of Ca2+ from ER is inefficient. It is very likely that endothelial [Ca2+]m is insensitive to Ca2+ mobilization by IP3 because mitochondria in EC are not closely associated with ER: <4% of mitochondria are within 700 nm of the ER in immortalized human umbilical vein endothelial cells. It appears that it is an influx of extracellular Ca2+ in EC that generates a microdomain of high concentration of Ca2+ close to the plasma membrane causing an elevation of Ca2+ in mitochondria located in this region (Fig. 7.1). Different from ECs, other nonexcitable cells (e.g., HeLa, hepatocytes) contain majority of mitochondria in a close proximity to ER [5] and demonstrate rapid transient elevation in [Ca2+]m sensitive to IP3-dependent Ca2+ mobilization [7–9].
The role of mitochondria as energy sources in EC might become significant in response to the metabolic disturbance associated with diabetes. Under these conditions, activation of endothelial fuel-sensing AMP kinase promotes oxidation of fatty acids (FAs) in mitochondria as a source of ATP production, whereas dependence of the cell on glycolysis decreases [10].
Role of Endothelial Mitochondria in the Generation of Reactive Oxygen Species
Considerable evidence supports a causal role for ROS produced by EC. ROS act as cell-signaling molecules, and their enhanced production directly inactivates endothelium-derived NO and promotes protein dysfunction—events that contribute to the initiation and progression of endothelial dysfunction. Endothelial mitochondria are one of the major enzymatic sources responsible for the production of ROS and elevation of them noted in vascular disease (other sources are NADPH oxidase, xanthine oxidase, and “uncoupled” eNOS). Production of highly reactive ROS, superoxide anion (O2•−), can occur during normal respiration at both complex I (partial reduction of molecular O2) and complex III (production of semiquinone radical) in mitochondria isolated from bovine aortic endothelial cells [11]. Complex III has been described as the main site of ROS production in human umbilical vein endothelial cells during hypoxia-reoxygenation [12] and after stimulation with TNF-α [13]. In human coronary arteriolar endothelial cells, complex I- and/or III-generated ROS participate in dilation in response to shear stress [14]. Mitochondrial generation of O2•− represents a major intracellular source of ROS under physiologic conditions. The levels of mitochondrial O2•− are influenced by the local O2 concentration, efficiency of mitochondrial ETC, the activities of mitochondrial manganese superoxide dismutase (MnSOD; located in the matrix of the organelle), and uncoupling proteins (UCPs).
Dismutation of O2•− by MnSOD results in the formation of another ROS, the H2O2. The latter mediates redox signaling and is regulated by mitochondrial thioredoxin, glutaredoxin, and peroxiredoxin (see below). ROS regulate Ca2+-handling proteins (ryanodine receptor, sarco-/endoplasmic reticulum calcium ATPase (SERCA), mitochondrial Na+/Ca2+ exchanger) [15] and mitochondrial electron transport chain itself. Low concentrations of ROS or RNS are now recognized as physiologic intracellular messengers. For instance, removal of endogenous H2O2 inhibits EC proliferation and angiogenesis [16, 17]. Conversely, low concentrations of exogenous H2O2 enhance proliferation of cultured human EC or tube formation by bovine aortic EC [18, 19]. Mitochondria can sense and transduce the redox signal: in cells deficient in functional mitochondria or exposed to inhibitors of the mitochondrial ETC, H2O2-induced transactivation of growth-factor receptors (EGF, PDGF, VEGF receptors) and resultant stimulation of JNK, p53, and Akt are attenuated (Fig. 7.2) [20].
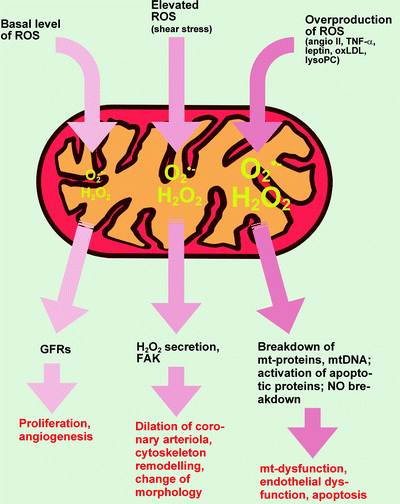
Fig. 7.2
Mitochondria-derived reactive oxygen species in the endothelial cell as a physiologic intracellular messengers and a source of an oxidative damage. See text for details. Abbreviations: angio II angiotensin II; FAK focal adhesion kinase; GFRs growth factor receptors; H 2 O 2 hydrogen peroxide; lysoPC lysophosphatidylcholine; mt mitochondrial; NO nitric oxide; O2•− superoxide anion; oxLDL oxidized low-density lipoprotein; ROS reactive oxygen species; TNF-α, tumor necrosis factor-α
Optimal redox signaling is controlled by antioxidant networks. Several antioxidant enzymes reside in mitochondria: glutathione peroxidase (Gpx) 1, peroxiredoxin (Prx) 3, oxidoreductase thioredoxin (Trx) 2, and glutathione-dependent oxidoreductase glutaredoxin (Grx) 2. A role of mitochondrial Prx3 for control of local H2O2 concentration was revealed by a study showing that depletion of Prx3 enhanced mitochondrial H2O2 production and apoptosis in cells stimulated with TNF-α [21]. Antiapoptotic function of Trx2 is based on its interaction with and inhibition of mitochondrial proapoptotic apoptosis signal-related kinase-1 (ASK1) [22].
Various physiologic and pathogenic stimuli can promote the formation of O2•− in mitochondria of vascular endothelium. For example, angiotensin II-mediated activation of NF-κB, VCAM-1 expression [23], and TNF-α-induced cytotoxicity [24, 25] involves generation of mitochondrial ROS. Interestingly, adipokine leptin stimulates O2•− production in mitochondria of aortic ECs via PKA-dependent increase of free fatty acid (FFA) oxidation [26].
In the endothelium of human coronary arterioles, generation of H2O2 in mitochondria is induced by shear stress. This H2O2 mediates flow-induced dilation of vessels via diffusion to the underlying vascular smooth muscle cells and activation of large-conductance Ca2+-activated potassium channels [14]. One potential mechanism of shear stress-induced release of ROS from mitochondria may involve cytoskeletal microfilaments that functionally connect sarcolemmal and mitochondrial membranes in EC either directly or indirectly. Within the EC, mechanic stimuli-induced mitochondrial ROS initiate wide range of responses including changes in gene expression, remodeling of cytoskeletal organization, and cellular morphology [27]. Recent studies have indicated that at least in pulmonary arterial ECs, mitochondrial ROS activate PKC. PKC phosphorylates/activates focal adhesion kinase (FAK) involved in a wide range of responses in ECs (Fig. 7.2) [28, 29].
ROS signaling would be disrupted by oxidative stress when huge quantities of ROS are generated. Thus, exposure of EC to H2O2, oxidized low-density lipoprotein (oxLDL), and lysophosphatidylcholine (lysoPC) promotes mitochondria-dependent ROS production (Fig. 7.2) [30–32]. The mechanism of this process appears to involve an interplay between the Ca2+-dependent mitochondrial dehydrogenases and ETC complex I and lead to the selective activation the Erk/MAPK pathway [33–35]. According to Chandel et al. [36] ETC complex III is involved in hypoxia-induced mitochondrial ROS generation and decreased AP-1 transcriptional activity. One of the consequences of mitochondrial ROS generation by ECs at low O2 concentrations is phosphorylation/activation of AMP-activated protein kinase (AMPK) [37]. It might indicate cell defense mechanism, as AMPK-dependent cardioprotection and the inactivation of caspase-3 in ECs have been demonstrated [38, 39].
Recent studies highlighted a role of PKCβ and 66-kDa adaptor protein Shc (p66Shc) in the vascular complications. Thus, inhibition of PKCβ improves function of endothelium under hyperglycemic conditions [40]; deficiency in p66Shc is protective against ROS- and aging-induced endothelial dysfunction [41, 42]. It appears that in response to oxidative stress, activated PKCβ phosphorylates p66Shc. Phosphorylated p66Shc translocates to the mitochondria where it interferes with ETC through oxidation of cytochrome c to generate ROS and trigger mitochondrial apoptosis [43, 44].
Abnormal overproduction of mitochondrial ROS leads to an oxidative damage of mitochondrial proteins, lipids, and nucleic acid, leading eventually to mitochondrial dysfunction. In addition, rapid interaction of O2•− with NO directly inactivates this vasodilator and simultaneously forms another oxidant, peroxynitrite (ONOO−), which can oxidize and uncouple NOS and further enhance O2•− production. This is why stimuli raising mitochondrial O2•− (angiotensin II, CD40) decrease NO bioactivity in aortic EC [45, 46]. ROS (peroxynitrite, H2O2) irreversibly damage proteins of the electron transport chain [47], raise mitochondrial Ca2+ via inactivation of mitochondrial Na+/Ca2+ exchanger [48], and damage mtDNA, resulting in decreased mitochondrial protein synthesis and mitochondrial dysfunction (Fig. 7.2) [49].
Enhanced mitochondrial O2•− flux in ECs occurs under hyperglycemic conditions, which develop in patients with type 2 diabetes, and this elevated O2•− has been implicated in glucose-mediated vascular damage [50–52]. It has been shown that mitochondrial excess of O2•− in ECs inhibits a glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), resulting in the activation of the major pathways of hyperglycemic damage. The following mechanism underlies this process: O2•− produces nuclear DNA strand breaks, which activate poly(ADP ribose) polymerase. The latter enzyme poly(ADP-ribosyl)ates and inhibits GAPDH. Inhibition of GAPDH increases the entry of upstream glycolytic metabolites into pathways of glucose overuse (formation of advanced glycation end products and PKC glucotoxic pathways) [51–53]. One of the complications found in insulin-resistant subjects is increased levels of FFAs. It has been shown that high concentrations of FFAs increase O2•− production in EC several fold via affecting mitochondrial ETC [54], and this leads to dysfunction of ECs, increased subendothelial LDL oxidation, and activation of hexosamine, PKC, and AGE pathways—factors that promote atherosclerosis [54–56].
Role of Endothelial Mitochondria in the Generation of NO
Endothelial mitochondria take part in the production of NO. An existence of mitochondrial NO synthase (mtNOS) is well documented. Brodsky et al. [57] demonstrated local NO generation in mitochondria of EC and in isolated mitochondria. Production of NO by mitochondria was stimulated by Ca2+ and was sensitive to NOS inhibitor, nitroarginine (L-NNA). The necessity of mitochondrial Ca2+ elevation for maximal mtNOS activity in EC was confirmed by others [58]. Dependence of mitochondrial NO production on Ca2+ [57, 58] and calmodulin [58] argues for endothelial isoform of NOS (eNOS) being present and synthesizing NO in endothelial mitochondria. In the strong agreement with this suggestion, immunodetection reveals eNOS bound to the mitochondrial outer membrane (MOM) of human umbilical vein EC [57] and calf pulmonary artery EC [58]. Mitochondrial Ca2+ accumulation significantly increases mtNOS phosphorylation in vascular EC at Ser-1177, suggesting Akt-dependent phosphorylation/activation of mtNOS during stimulation of mitochondrial Ca2+ uptake [58].
One potential role of mtNOS is the regulation of the electron transport along with the respiratory chain in endothelial mitochondria via a reversible, competitive inhibition of cytochrome c oxidase (complex IV) activity. More specifically, NO reduces the affinity of cytochrome c oxidase for O2, and this can lead to the changes in the cellular O2 consumption and availability of O2 for other cellular processes. In addition, NO exerts its inhibitory effects on respiratory complex III as well, by inhibiting electron transfer between cytochromes b and c 1.
Аctivation of NO production in endothelial mitochondria in response to Ca2+ uptake demonstrates a negative feedback regulation: decrease of mitochondrial Ca2+ uptake and, hence, reduced mtNOS activity in vascular EC. This mechanism may be particularly important to protect against mitochondrial Ca2+ overload under pathological conditions when cellular concentration of NO can reach very high levels. The protection is mediated by NO-dependent depolarization of mitochondrial membrane, which leads to the decreased mitochondrial Ca2+ uptake through the electrogenic mitochondrial Ca2+ uniporter. An additional emergency mechanism preventing mitochondrial Ca2+ overload is the reversible NO-driven opening of the mitochondrial permeability transition pore (MPTP) leading to fast release of accumulated Ca2+ ions [59].
Mitochondria-specific NOS can also provide a fast local modulatory effect of NO on cell apoptosis. Data of Ghafourifar et al. [60] confirmed that Ca2+ uptake by mitochondria from rat liver triggers mtNOS activity and stimulates apoptosis (Bcl-2-sensitive cytochrome c release from mitochondria, an increase of lipid peroxidation) via formation of powerful oxidizing agent, peroxynitrite, from NO and O2•−. Although there is no direct evidence for NO-induced apoptosis in ECs, data of Dedkova et al. [58] suggest that part of the NO formed by mtNOS in ECs is quickly transformed into peroxynitrite, which can further induce apoptosis.
In EC subjected to the hyperglycemic microenvironment (high concentration of d-glucose), mtNOS becomes “uncoupled”: it switches from a NO-generating to a superoxide-producing enzyme [57]. Thus, malfunctioning mtNOS contributes to the hyperglycemia-caused oxidant stress in EC and as such could be responsible for diverse metabolic derangements occurring in the vascular endothelium.
Endothelial Mitochondria and Apoptosis
Development of several diseases (atherosclerosis, diabetes, heart failure, ischemia) is related to impairment of vascular function caused by oxidative stress. Vascular ECs as the frontline against these disturbances are critical targets of oxidative stress. Opening of the mitochondrial permeability transition pore and activation of mitochondrial pathways of apoptosis are important factors that compromise survival of endothelial cells and contribute to development of abovementioned diseases.
The MPTP is a large, nonspecific pore thought of spanning the inner and outer mitochondrial membranes. Some components of MPTP include the adenine nucleotide translocator, the voltage-dependent anion channel, and cyclophilin D in the matrix (see Chap. 3). In cells, an increase in free Ca2+, a decrease in mitochondrial membrane potential, ATP depletion, increased phosphate levels, increased acidity, and oxidative stress cause opening of MPTP, leading to necrosis (Fig. 7.3). Oxidative stress-dependent MPTP opening may contribute to the EC death in diabetes. For example, oxidative stress or hyperglycemia has been found to open MPTP in both HUVECs and bovine aortic EC [61] and induce apoptosis of human aortic EC via mitochondrial depolarization/MPTP opening [62]. Interestingly, MPTP in proliferating EC opens easier than in quiescent ECs [63], and this phenomenon can be used to selectively kill angiogenic EC to inhibit tumor angiogenesis.
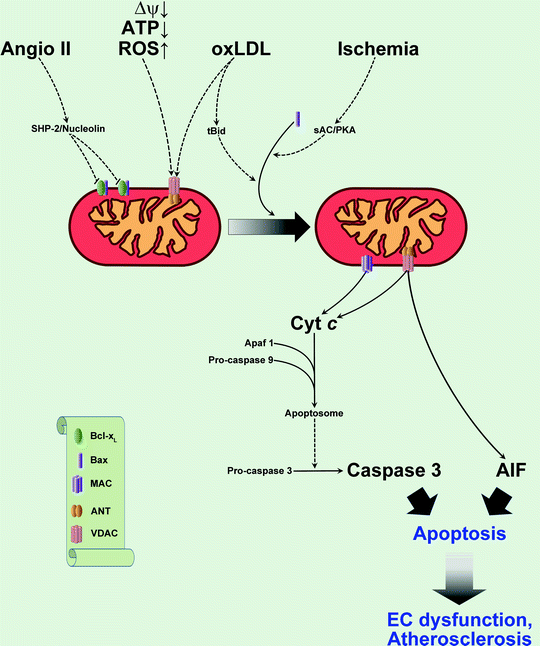




Fig. 7.3
Mitochondria-related apoptotic pathways in the vascular endothelium. Proatherogenic factors [oxidized low-density lipoprotein (oxLDL), angiotensin II (Angio II), oxidative stress] and ischemia activate intrinsic apoptotic pathways in endothelial cells (see text for details) leading to the release of proapoptotic factors from mitochondria [cytochrome c (Cyt c), apoptosis-inducing factor (AIF)] via mitochondrial apoptosis-induced channel (MAC) and mitochondrial permeability transition pore (MPTP). As a result, apoptosis of endothelial cells contributes to development of disease. For simplification, only two components of MPTP are shown, voltage-dependent anion channel (VDAC) and adenine nucleotide translocator (ANT). Other abbreviations: Apaf 1 apoptotic protease-activating factor 1; Bcl-x L B-cell lymphoma-extra large protein; ΔΨ mitochondrial membrane potential; EC endothelial cell; PKA protein kinase A; ROS reactive oxygen species; sAC soluble adenylate cyclase; SHP-2 Src homology 2-containing protein tyrosine phosphatase; tBid truncated BH3-interacting domain death agonist. Translocations of molecules are shown by solid arrows; regulatory effects are shown by dashed arrows
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
