Extended-release niacin (ERN) improves multiple lipid parameters but is underused owing to niacin-induced flushing (NIF). Laropiprant (LRPT) reduces NIF; however, its effects on chronic flushing (>6 months) have not been studied. We examined whether after 20 weeks of treatment with ERN/LRPT, patients who continued ERN/LRPT would experience less NIF than patients who stopped LRPT and continued ERN alone. A total of 1,152 dyslipidemic patients were randomized 2:2:1 to group 1, ERN/LRPT 1 g/20 mg/day from 0 to 4 weeks and then ERN/LRPT 2 g/40 mg/day from 5 to 32 weeks; group 2, ERN/LRPT 1 g/20 mg/day from 0 to 4 weeks, ERN/LRPT 2 g/40 mg/day from 5 to 20 weeks, and then ERN 2 g/day without LRPT from 21 to 32 weeks; or group 3, placebo for the entire study. The end points included the number of days each week with a moderate or greater Global Flushing Severity Score (GFSS) ≥4 (primary end point) and the percentage of patients with a maximum GFSS of ≥4 (secondary end point) during the postwithdrawal period (weeks 21 to 32). ERN/LRPT produced significantly less NIF than ERN alone during the postwithdrawal period, as measured by the number of days each week with a GFSS of ≥4 (p <0.001) and the percentage of patients with a maximum GFSS of ≥4 (p <0.001; ERN/LRPT 19.6%; ERN 48.9%; placebo 9.2%). Compared with ERN alone, ERN/LRPT produced fewer drug-related adverse experiences during the postwithdrawal period. After 20 weeks of stable maintenance therapy, dyslipidemic patients treated continuously with ERN/LRPT experienced less NIF than did patients who had had LRPT withdrawn and had continued with ERN alone. In conclusion, the results of our study support the long-term efficacy of ERN/LRPT in reducing NIF symptoms.
Niacin is underused owing to niacin-induced flushing (NIF) of the face and trunk that occurs in >90% of patients taking niacin. NIF is mediated primarily by prostaglandin D 2 , which stimulates prostaglandin D 2 receptor 1 in the skin. Laropiprant (LRPT) is a potent, once-daily, highly selective prostaglandin D 2 receptor 1 antagonist. We have previously demonstrated that a fixed-dose combination tablet of extended-release niacin/laropiprant (ERN/LRPT) improved low-density lipoprotein (LDL) cholesterol, high-density lipoprotein cholesterol, triglycerides, and other atherogenic lipids and reduced the incidence, frequency, and intensity of NIF compared with ERN alone for ≤6 months in patients with primary hypercholesterolemia or mixed dyslipidemia ( Figure 1 ). To examine the effectiveness of LRPT beyond 6 months of ERN/LRPT treatment, the present Phase III, randomized placebo-controlled clinical trial tested the hypothesis that after 20 weeks of ERN/LRPT, patients who continued taking ERN/LRPT would experience less NIF than patients who had LRPT withdrawn and continued with ERN alone.
Methods
The present study enrolled dyslipidemic men and women aged 18 to 75 years, who were appropriate for lipid-altering therapy as determined by the study investigators. The lipid entry criteria were determined from the National Cholesterol Education Program Adult Treatment Panel III risk categorization. Thus, patients with a high risk of cardiovascular disease (coronary heart disease or coronary heart disease risk equivalent) were required either to be taking a statin and have LDL cholesterol <100 mg/dl (<2.59 mmol/L) or be intolerant to statins and have LDL cholesterol <120 mg/dl (<3.11 mmol/L). The patients with multiple cardiovascular disease risk factors (≥2 risk factors and Framingham 10-year coronary heart disease risk of ≤20%) were required to have LDL cholesterol <130 mg/dl (<3.37 mmol/L). Finally, patients with low cardiovascular disease risk (0 to 1 risk factor) were required to have LDL cholesterol <190 mg/dl (<4.92 mmol/L). In addition, all patients had to have triglycerides <500 mg/dl (<5.65 mmol/L).
Patients were excluded if they had the following laboratory values at visit 1: creatinine >2.0 mg/dl (177 μmol/L), alanine aminotransferase >1.5 times the upper limit of normal (ULN), aspartate aminotransferase >1.5 times the ULN, and creatine kinase >2 times the ULN. Patients were also excluded if they were currently experiencing menopausal hot flashes. Patients with poorly controlled (glycosylated hemoglobin >8.5% at visit 1) type 1 or type 2 diabetes mellitus were also excluded. Patients were ineligible if they were taking niacin >50 mg/day or fibrates and statins concomitantly. Because of the potential effects on NIF, aspirin >100 mg/day and long-acting nonsteroidal anti-inflammatory drugs were also excluded.
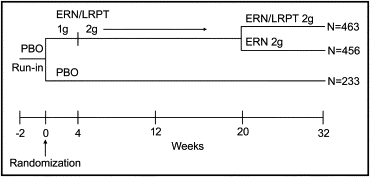
The present study was a multicenter, randomized, double-blind, placebo-controlled study that, after a 2-week placebo run-in period, included a 32-week double-blind treatment period. Patients without previous lipid-altering drug therapy were eligible to participate, as were those with previous lipid-altering therapy, as long as such therapy had been stable for 6 weeks before the placebo run-in period. The eligible patients were randomized in a 2:2:1 ratio to 1 of 3 treatment groups: group 1, ERN/LRPT 1 g/20 mg/day from 0 to 4 weeks and then ERN/LRPT 2 g/40 mg/day for the remainder of the study (ERN/LRPT to ERN/LRPT); group 2, ERN/LRPT 1 g/20 mg/day from 0 to 4 weeks, ERN/LRPT 2 g/40 mg/day from 5 to 20 weeks, and then ERN 2 g/day from 21 to 32 weeks (ERN/LRPT to ERN); or group 3, placebo for the duration of the study ( Figure 2 ). Randomization of the study drug was achieved using an interactive voice response system. The patients were instructed to take the study therapy with food in the evening or at bedtime. To reduce the possibility of NIF symptoms (skin redness, feeling warm, or tingling), patients were advised to avoid alcohol, spicy foods, and hot drinks around the time of dosing. Nine scheduled clinic visits included weeks −3, −2, 0 (day 1), 4, 10, 16, 20, 26, and 32. The final visit was followed 14 days later by a poststudy telephone interview to assess for potential serious adverse experiences (AEs).
Patients were asked to complete a previously validated Flushing Symptom Questionnaire (FSQ), provided through an electronic diary (e-diary), that included 11 questions that assessed aspects of the frequency, severity, duration, and bother of flushing. The patients used a hand-held e-diary to ensure the completeness and reliability of the patient-reported flushing data. The e-diary was completed each morning and was intended to reflect flushing symptoms experienced during the previous day. One of the FSQ items assessed the intensity of all 4 flushing symptoms (skin redness, warmth, tingling, and/or itching) in aggregate using an 11-point numeric rating scale. This was termed the Global Flushing Severity Score (GFSS), which was labeled in the e-diary with the following intensity categories: none (0), mild (1 to 3), moderate (4 to 6), severe (7 to 9), and extreme (10). Patients were followed up closely (daily uploading of the e-diary, frequent clinic visits, and drug accountability) to ensure daily compliance.
The appropriate ethics committees/institutional review boards approved the study protocol. All patients provided written informed consent. The study was performed under the guidelines established by the Declaration of Helsinki and Good Clinical Practice standards. The clinical trial number was NCT00961636 .
The primary end point was the number of days each week with moderate or greater flushing (GFSS ≥4) across the postwithdrawal period (weeks 21 to 32) and was categorized as 0, >0 to 0.5, >0.5 to 1, >1 to 2, >2 to 3, and >3 days/week. The secondary end point was the percentage of patients with moderate or greater flushing (GFSS ≥4) across the postwithdrawal period. The exploratory flushing end points included the biweekly maximum GFSS averaged across the postwithdrawal period and the weekly maximum GFSS averaged across the postwithdrawal period.
The safety assessments included physical examination, vital signs, laboratory evaluations, AE monitoring, and patient interviews. An AE was considered “drug-related” if it was determined by the investigator to be possibly, probably, or definitely due to study drug. The prespecified safety parameters of interest included hepatitis-related clinical AEs (according to a predefined set of “Medical Dictionary for Regulatory Activities” terms), consecutive elevations in alanine aminotransferase/aspartate aminotransferase ≥3 times the ULN, single elevations in the alanine aminotransferase/aspartate aminotransferase ≥5 times the ULN and ≥10 times the ULN, creatine kinase elevations of ≥10 times the ULN (any) and ≥10 times the ULN with drug-related muscle symptoms (definition of myopathy), confirmed adjudicated cardiovascular events, and clinically recognized new diabetes (defined as experiencing an AE related to the diagnosis of diabetes mellitus or the initiation of antidiabetic medication).
The full analysis set population was used for the primary efficacy end points and included all randomized patients who had a withdrawal visit (visit 7) and ≥1 GFSS score during the postwithdrawal period (weeks 21 to 32). The primary hypothesis regarding flushing was assessed by comparing the ERN/LRPT and ERN groups for the primary end point (number of days each week with moderate or greater GFSS [GFSS ≥4] across the postwithdrawal period [weeks 21 to 32]. The comparison was performed using the Cochran-Mantel-Haenszel test stratified by country applied to these 2 groups, with the number of days categorized as 0, >0 to 0.5, >0.5 to 1, >1 to 2, >2 to 3, and >3 days/week. The secondary end point (percentage of patients with moderate or greater GFSS [GFSS ≥4] across the postwithdrawal period [weeks 21 to 32]) in the ERN/LRPT and ERN groups was compared using the unconditional Miettinen and Nurminen method stratified by country.
All randomized patients who had received ≥1 dose of the treatment period drug were included in the safety analyses. For the prewithdrawal (weeks 1 to 20) and postwithdrawal (weeks 21 to 32) periods, statistical tests were performed, and the 95% CI and p values are provided for the prespecified safety parameters of interest. All serious AEs that occurred within 14 days of the last dose of the study drug (after completion or discontinuation of the study) were collected. For patients who withdrew from the study, an additional poststudy safety follow-up assessment of serious cardiovascular events and deaths was conducted on the date of the originally planned end-of-study visit (32 weeks after randomization). Those serious AEs and adjudicated cardiovascular events occurring within the 14-day follow-up window were included in the safety analyses.
Results
The demographics and baseline characteristics were generally similar across the treatment groups ( Table 1 ). Of the 1,152 patients randomized, 1,148 received the study drug and 270 (23.4%) discontinued before completing the study. Of the 270 patients who discontinued, 32 (13.7%) were in the placebo, 131 (28.7%) were in the ERN/LRPT to ERN, and 107 (23.1%) were in the ERN/LRPT to ERN/LRPT group.
| Variable | ERN/LRPT to ERN/LRPT (n = 463) | ERN/LRPT to ERN (n = 456) | Placebo (n = 233) |
|---|---|---|---|
| Men | 60.3% | 68.4% | 62.7% |
| White | 92.2% | 92.3% | 92.3% |
| Age (years) | |||
| Mean | 58.3 | 57.8 | 58.0 |
| Range | 24–75 | 18–76 | 22–75 |
| Patients aged ≥65 years | 27.9% | 28.1% | 28.8% |
| Statin use ⁎ | 59.6% | 63.4% | 58.8% |
| Previous niacin use † | 13.6% | 14.3% | 12.9% |
| Diabetes mellitus | 22.0% | 23.2% | 20.6% |
| High coronary heart disease risk ‡ | 33.3% | 36.2% | 29.2% |
⁎ As background lipid-modifying therapy during study.
† Discontinued ≥6 weeks before visit 1.
‡ Per National Cholesterol Education Program Adult Treatment Panel III.
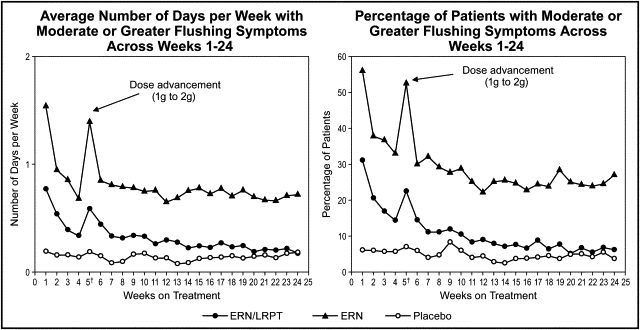
During the postwithdrawal period (weeks 21 to 32), ERN/LRPT produced significantly (p <0.001) less flushing relative to ERN, as measured by the number of days each week with moderate or greater flushing (GFSS ≥4), categorized as 0, >0 to 0.5, >0.5 to 1, >1 to 2, >2 to 3, and >3 days/week (p <0.001; primary end point) and the percentage of patients with the maximum GFSS ≥4 (p <0.001; secondary end point; Figures 3 and 4 ). During the postwithdrawal period, there was also less flushing in the ERN/LRPT treatment group than in the ERN treatment group, as measured by the average of the biweekly maximum GFSS (p <0.001) and the average of the weekly maximum GFSS (p <0.001; data not shown).



