EDEMA
Eugene Braunwald ![]() Joseph Loscalzo
Joseph Loscalzo
Edema is defined as a clinically apparent increase in the interstitial fluid volume, which may expand by several liters before the abnormality is evident. Therefore, a weight gain of several kilograms usually precedes overt manifestations of edema, and a similar weight loss from diuresis can be induced in a slightly edematous patient before “dry weight” is achieved. Anasarca refers to gross, generalized edema. Ascites and hydrothorax refer to accumulation of excess fluid in the peritoneal and pleural cavities, respectively, and are considered special forms of edema.
Depending on its cause and mechanism, edema may be localized or have a generalized distribution. Edema is recognized in its generalized form by puffiness of the face, which is most readily apparent in the periorbital areas, and by the persistence of an indentation of the skin after pressure; this is known as “pitting” edema. In its more subtle form, edema may be detected by noting that after the stethoscope is removed from the chest wall, the rim of the bell leaves an indentation on the skin of the chest for a few minutes. When the ring on a finger fits more snugly than in the past or when a patient complains of difficulty putting on shoes, particularly in the evening, edema may be present.
PATHOGENESIS
About one-third of total-body water is confined to the extracellular space. Approximately 75% of the latter is interstitial fluid, and the remainder is in the plasma compartment.
Starling forces
The forces that regulate the disposition of fluid between these two components of the extracellular compartment frequently are referred to as the Starling forces. The hydrostatic pressure within the vascular system and the colloid oncotic pressure in the interstitial fluid tend to promote movement of fluid from the vascular to the extravascular space. By contrast, the colloid oncotic pressure contributed by plasma proteins and the hydrostatic pressure within the interstitial fluid promote the movement of fluid into the vascular compartment.
As a consequence of these forces, there is movement of water and diffusible solutes from the vascular space at the arteriolar end of the capillaries. Fluid is returned from the interstitial space into the vascular system at the venous end of the capillaries and by way of the lymphatics. Unless these channels are obstructed, lymph flow rises with increases in net movement of fluid from the vascular compartment to the interstitium. These flows are usually balanced so that there is a steady state in the sizes of the intravascular and interstitial compartments, yet a large exchange between them occurs. However, if either the hydrostatic or the oncotic pressure gradient is altered significantly, a further net movement of fluid between the two components of the extracellular space will take place. The development of edema then depends on one or more alterations in the Starling forces so that there is increased flow of fluid from the vascular system into the interstitium or into a body cavity.
Edema due to an increase in capillary pressure may result from an elevation of venous pressure caused by obstruction to venous and/or lymphatic drainage. An increase in capillary pressure may be generalized, as occurs in congestive heart failure (see later). The Starling forces also may be imbalanced when the colloid oncotic pressure of the plasma is reduced owing to any factor that may induce hypoalbuminemia, such as severe malnutrition, liver disease, loss of protein into the urine or into the gastrointestinal tract, or a severe catabolic state. Edema may be localized to one extremity when venous pressure is elevated due to unilateral thrombophlebitis (see later).
Capillary damage
Edema may also result from damage to the capillary endothelium, which increases its permeability and permits the transfer of proteins into the interstitial compartment. Injury to the capillary wall can result from drugs, viral or bacterial agents, and thermal or mechanical trauma. Increased capillary permeability also may be a consequence of a hypersensitivity reaction and is characteristic of immune injury. Damage to the capillary endothelium is presumably responsible for inflammatory edema, which is usually nonpitting, localized, and accompanied by other signs of inflammation—i.e., erythema, heat, and tenderness.
Reduction of effective arterial volume
In many forms of edema, the effective arterial blood volume, a parameter that represents the filling of the arterial tree, is reduced. Underfilling of the arterial tree may be caused by a reduction of cardiac output and/or systemic vascular resistance. As a consequence of underfilling, a series of physiologic responses designed to restore the effective arterial volume to normal are set into motion. A key element of these responses is the retention of salt and, therefore, of water, ultimately leading to edema.
Renal factors and the renin-angiotensin-aldosterone (RAA) system
In the final analysis, renal retention of Na+ is central to the development of generalized edema (Fig. 7-1). The diminished renal blood flow characteristic of states in which the effective arterial blood volume is reduced is translated by the renal juxtaglomerular cells (specialized myoepithelial cells surrounding the afferent arteriole) into a signal for increased renin release. Renin is an enzyme with a molecular mass of about 40,000 Da that acts on its substrate, angiotensinogen, an α2-globulin synthesized by the liver, to release angiotensin I, a decapeptide, which in turn is converted to angiotensin II (AII), an octapeptide. AII has generalized vasoconstrictor properties; it is especially active on the renal efferent arterioles. This action reduces the hydrostatic pressure in the peritubular capillaries, whereas the increased filtration fraction raises the colloid osmotic pressure in these vessels, thereby enhancing salt and water reabsorption in the proximal tubule as well as in the ascending limb of the loop of Henle.
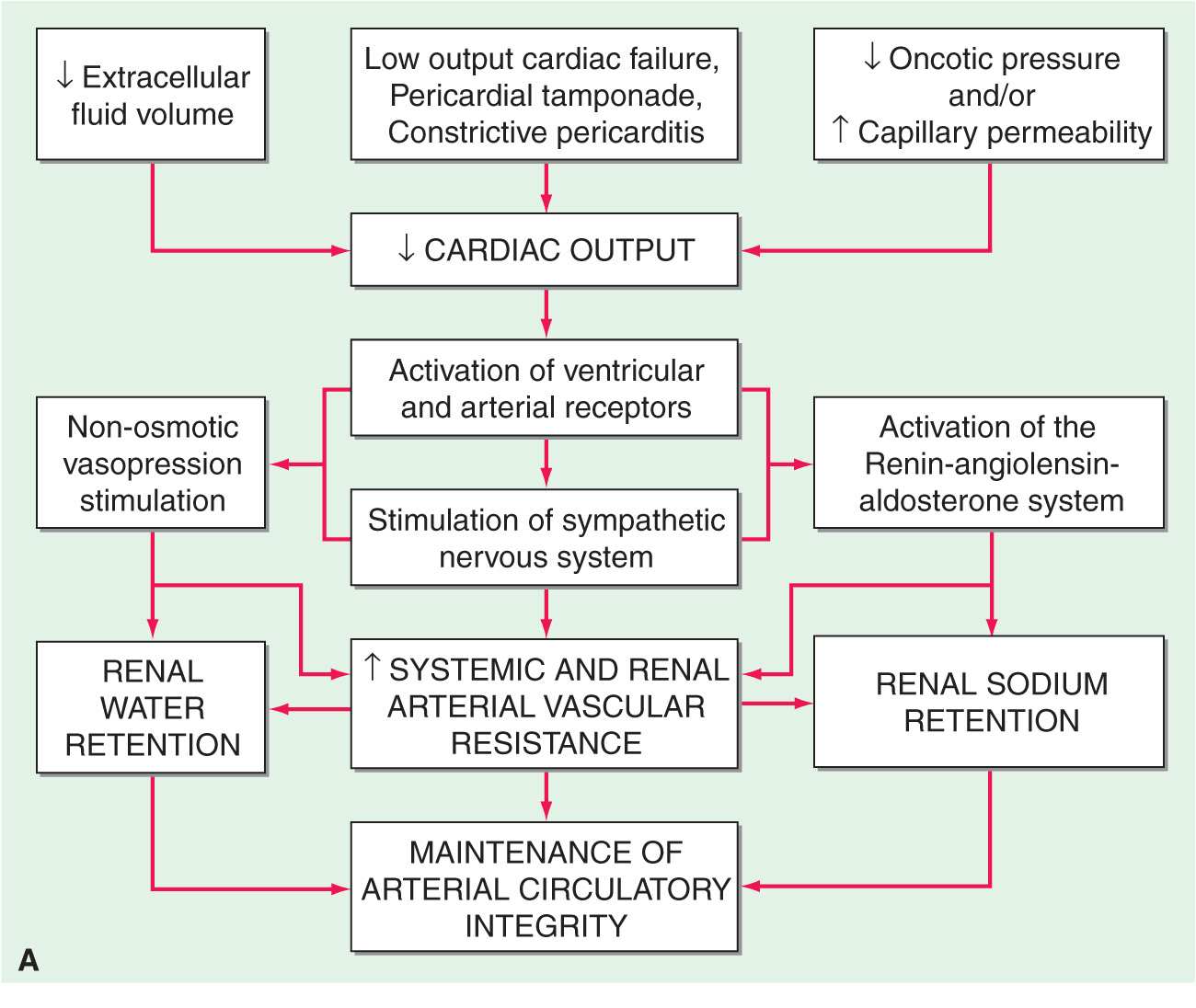
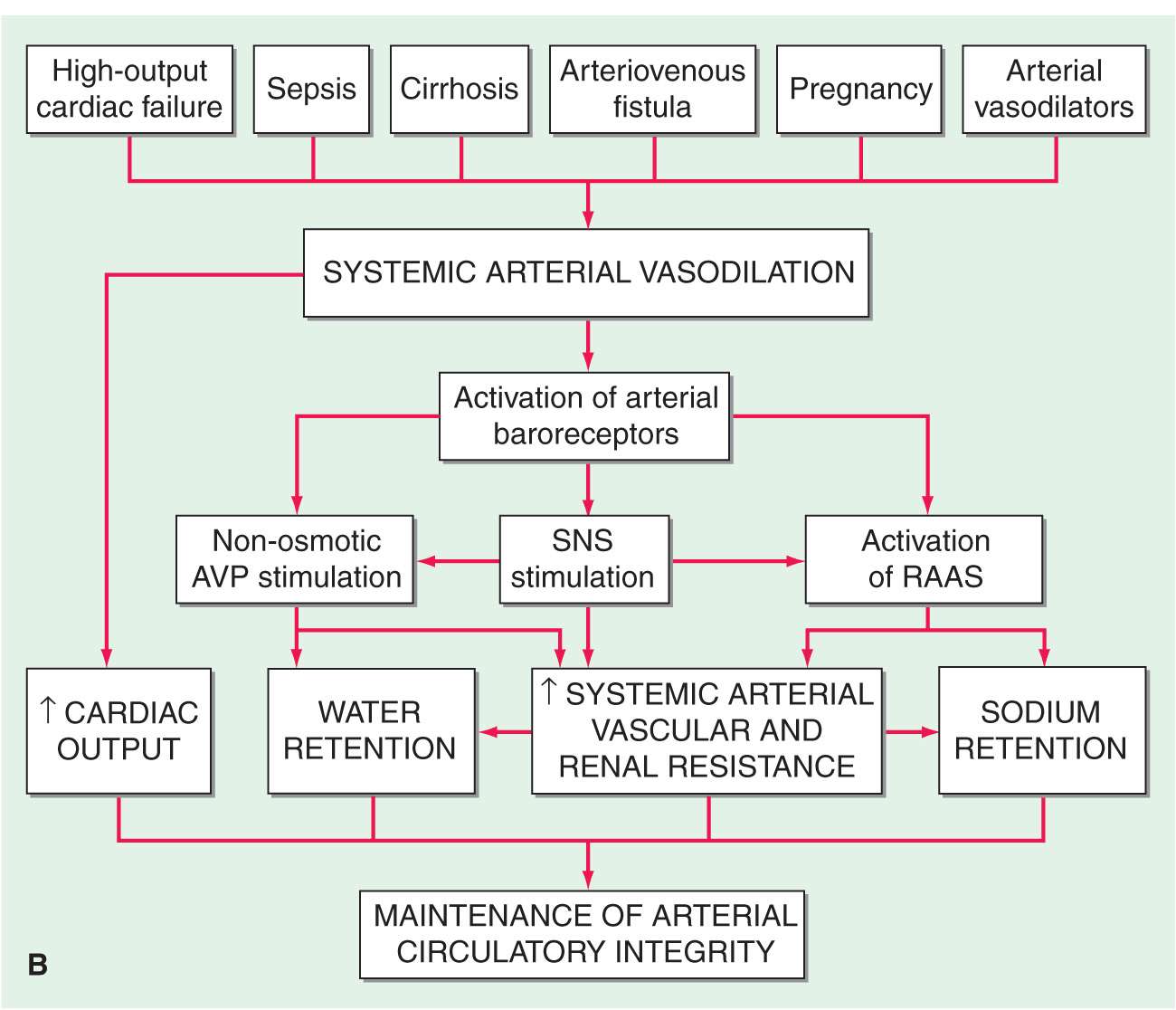
FIGURE 7-1
Clinical conditions in which a decrease in cardiac output (A) and systemic arterial vasodilation (B) cause arterial underfilling with resulting neurohumoral activation and renal sodium and water retention. In addition to activating the neurohumoral axis, adrenergic stimulation causes renal vasoconstriction and enhances sodium and fluid transport by the proximal tubule epithelium. SNS, sympathetic nervous system; RAAS, renin-angiotensin aldosterone system. (Reprinted from RW Schrier: Ann Intern Med 113:155, 1990.)
The renin-angiotensin-aldosterone (RAA) system has long been recognized as a hormonal system; however, it also operates locally. Intrarenally produced AII contributes to glomerular efferent arteriolar constriction, and this “tubuloglomerular feedback” causes salt and water retention and thereby contributes to the formation of edema.
AII that enters the systemic circulation stimulates the production of aldosterone by the zona glomerulosa of the adrenal cortex. Aldosterone in turn enhances Na+ reabsorption (and K+ excretion) by the collecting tubule. In patients with heart failure, not only is aldosterone secretion elevated but the biologic half-life of aldosterone is prolonged, which increases further the plasma level of the hormone. A depression of hepatic blood flow, especially during exercise, is responsible for reduced hepatic catabolism of aldosterone.
Increased quantities of aldosterone are secreted in heart failure and in other edematous states, and blockade of the action of aldosterone by spironolactone or eplerenone (aldosterone antagonists) or by amiloride (a blocker of epithelial Na+ channels) often induces a moderate diuresis in edematous states. Yet persistently augmented levels of aldosterone (or other mineralocorticoids) alone do not always promote accumulation of edema, as witnessed by the lack of significant fluid retention in most instances of primary aldosteronism. Furthermore, although normal individuals retain some NaCl and water with the administration of potent mineralocorticoids, such as deoxycorticosterone acetate and fludrocortisone, this accumulation is self-limiting despite continued exposure to the steroid, a phenomenon known as mineralocorticoid escape. The failure of normal individuals who receive large doses of mineralocorticoids to accumulate large quantities of extracellular fluid and develop edema is probably a consequence of an increase in glomerular filtration rate (pressure natriuresis) and the action of natriuretic substance(s) (see later). The continued secretion of aldosterone may be more important in the accumulation of fluid in edematous states because patients with edema secondary to heart failure, nephrotic syndrome, and hepatic cirrhosis are generally unable to repair the deficit in effective arterial blood volume. As a consequence, they do not develop pressure natriuresis.
Arginine vasopressin (AVP)
The secretion of AVP occurs in response to increased intracellular osmolar concentration, and, by stimulating V2 receptors, AVP increases the reabsorption of free water in the renal distal tubule and collecting duct, thereby increasing total-body water. Circulating AVP is elevated in many patients with heart failure secondary to a nonosmotic stimulus associated with decreased effective arterial volume. Such patients fail to show the normal reduction of AVP with a reduction of osmolality, contributing to edema formation and hyponatremia.
Endothelin
This potent peptide vasoconstrictor is released by endothelial cells. Its concentration is elevated in heart failure and contributes to renal vasoconstriction, Na+ retention, and edema in heart failure.
Natriuretic peptides
Atrial distention and/or a Na+ load cause release into the circulation of atrial natriuretic peptide (ANP), a polypeptide; a high-molecular-weight precursor of ANP is stored in secretory granules within atrial myocytes. Release of ANP causes (1) excretion of sodium and water by augmenting glomerular filtration rate, inhibiting sodium reabsorption in the proximal tubule, and inhibiting release of renin and aldosterone and (2) arteriolar and venous dilation by antagonizing the vasoconstrictor actions of AII, AVP, and sympathetic stimulation. Thus, ANP has the capacity to oppose Na+ retention and arterial pressure elevation in hypervolemic states.
The closely related brain natriuretic peptide (BNP) is stored primarily in ventricular myocardium and is released when ventricular diastolic pressure rises. Its actions are similar to those of ANP, and both BNP and ANP bind to the natriuretic receptor-A, which is found in the myocardium. Yet another natriuretic peptide, C-type (CNP), is of endothelial and renal origin. CNP binds preferentially to the natriuretic peptide receptor-B, which is expressed principally in veins. Circulating levels of ANP and BNP are elevated in congestive heart failure and in cirrhosis with ascites, but obviously not sufficiently to prevent edema formation. In addition, in edematous states there is abnormal resistance to the actions of natriuretic peptides.
CLINICAL CAUSES OF EDEMA
Obstruction of venous (and lymphatic) drainage of a limb
In this condition, the hydrostatic pressure in the capillary bed upstream (proximal) to the obstruction increases so that an abnormal quantity of fluid is transferred from the vascular to the interstitial space. Since the alternative route (i.e., the lymphatic channels) also may be obstructed or maximally filled, an increased volume of interstitial fluid in the limb develops (i.e., there is trapping of fluid in the interstitium of the extremity). The displacement of fluid into a limb may occur at the expense of the blood volume in the remainder of the body, thereby reducing effective arterial blood volume and leading to the retention of NaCl and H2O until the deficit in plasma volume has been corrected.
Congestive heart failure
(See also Chap. 17) In this disorder the impaired systolic emptying of the ventricle(s) and/or the impairment of ventricular relaxation promotes an accumulation of blood in the venous circulation at the expense of the effective arterial volume, and the aforementioned sequence of events (Fig. 7-1) is initiated. In mild heart failure, a small increment of total blood volume may repair the deficit of arterial volume and establish a new steady state. Through the operation of Starling’s law of the heart, an increase in ventricular diastolic volume promotes a more forceful contraction and may thereby maintain the cardiac output. However, if the cardiac disorder is more severe, fluid retention continues, and the increment in blood volume accumulates in the venous circulation, raising venous pressure and causing edema.
Incomplete ventricular emptying (systolic heart failure) and/or inadequate ventricular relaxation (diastolic heart failure) both lead to an elevation of ventricular diastolic pressure. If the impairment of cardiac function primarily involves the right ventricle, pressures in the systemic veins and capillaries rise, augmenting the transudation of fluid into the interstitial space and enhancing the likelihood of peripheral edema. The elevated systemic venous pressure is transmitted to the thoracic duct with consequent reduction of lymph drainage, further increasing the accumulation of edema. If the impairment of cardiac function involves the left ventricle primarily, pulmonary venous and capillary pressures rise. Pulmonary artery pressure rises, and this in turn interferes with the emptying of the right ventricle, leading to an elevation of right ventricular diastolic and central and systemic venous pressures, enhancing the likelihood of the formation of peripheral edema. The elevation of pulmonary capillary pressure may cause pulmonary edema, which impairs gas exchange. The resulting hypoxemia may impair cardiac function further, sometimes causing a vicious circle.
Nephrotic syndrome and other hypoalbuminemic states
The primary alteration in this disorder is a diminished colloid oncotic pressure due to losses of large quantities of protein into the urine. With severe hypoalbuminemia and the consequent reduced colloid osmotic pressure, the NaCl and H2O that are retained cannot be restrained within the vascular compartment, and total and effective arterial blood volumes decline. This process initiates the edema-forming sequence of events described earlier, including activation of the RAA system. Impaired renal function contributes further to the formation of edema. A similar sequence of events occurs in other conditions that lead to severe hypoalbuminemia, including (1) severe nutritional deficiency states, (2) severe, chronic liver disease (see later), and (3) protein-losing enteropathy.
Cirrhosis
This condition is characterized in part by hepatic venous outflow blockade, which in turn expands the splanchnic blood volume and increases hepatic lymph formation. Intrahepatic hypertension acts as a stimulus for renal Na+ retention and a reduction of effective arterial blood volume. These alterations frequently are complicated by hypoalbuminemia secondary to reduced hepatic synthesis, as well as systemic vasodilation. These effects reduce the effective arterial blood volume further, leading to activation of the RAA system, renal sympathetic nerves, and other NaCl- and H2O-retaining mechanisms. The concentration of circulating aldosterone often is elevated by the failure of the liver to metabolize this hormone. Initially, the excess interstitial fluid is localized preferentially proximal (upstream) to the congested portal venous system and obstructed hepatic lymphatics, i.e., in the peritoneal cavity (ascites). In later stages, particularly when there is severe hypoalbuminemia, peripheral edema may develop. The excess production of prostaglandins (PGE2 and PGI2) in cirrhosis attenuates renal Na+ retention. When the synthesis of these substances is inhibited by nonsteroidal anti-inflammatory drugs (NSAIDs), renal function deteriorates and Na+ retention increases.
Drug-induced edema
A large number of widely used drugs can cause edema (Table 7-1). Mechanisms include renal vasoconstriction (NSAIDs and cyclosporine), arteriolar dilation (vasodilators), augmented renal Na+ reabsorption (steroid hormones), and capillary damage (interleukin 2).
TABLE 7-1
DRUGS ASSOCIATED WITH EDEMA FORMATION

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


