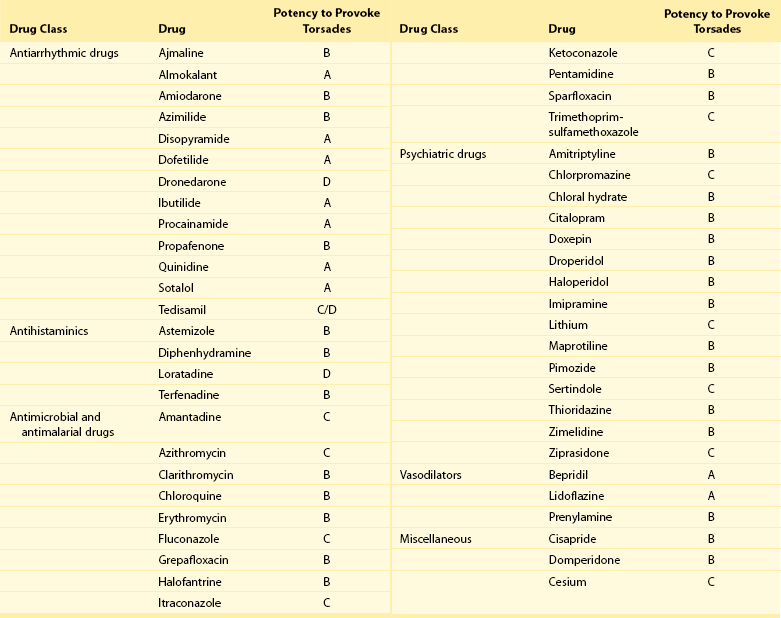
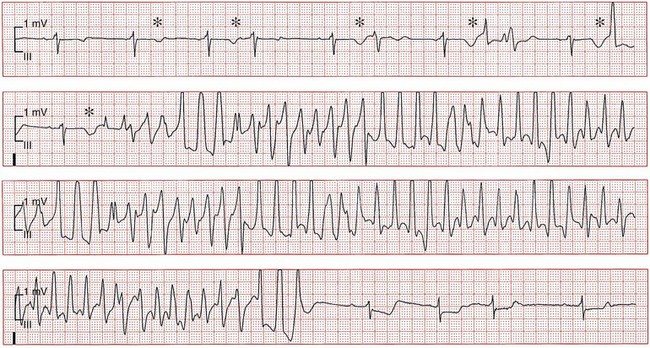
101 Drug-induced ventricular tachyarrhythmias can be caused by cardiovascular drugs, noncardiovascular drugs, and even nonprescription agents. They can also result in arrhythmic emergencies and sudden cardiac death.1 Development of a new arrhythmia, or aggravation of an existing arrhythmia, during therapy with a drug at a concentration usually considered not to be toxic is defined as proarrhythmia. This condition occurs in patients with or without structural heart disease. Major causes of proarrhythmia include direct pharmacologic effects on ion channels of the heart. Cardiac glycosides, QT-prolonging agents, and sodium channel–blocking drugs all have been implicated in this proarrhythmic category. In addition, certain drugs can cause a cardiomyopathy that ultimately leads to ventricular tachyarrhythmias—specifically, ventricular tachycardia. Others can cause coronary vasoconstriction and acute myocardial ischemia. The proportion of patients with proarrhythmia that initially manifests as sudden death, and the extent to which proarrhythmia contributes to the overall problem of sudden death, are unknown. Nevertheless, the experience with terfenadine or cisapride and the Cardiac Arrhythmia Suppression Trial (CAST)2 has illustrated the potential for drugs to result in unusual severe adverse effects, which can be difficult to detect. General treatment guidelines focus on avoidance of drug treatment in high-risk patients, recognition of the syndromes of drug-induced proarrhythmia, and withdrawal of the culprit agents. Specific therapies often are based on anecdotal evidence or experimental studies. The arrhythmogenic potential can manifest in patients who are especially sensitive to the electrophysiological effects of a particular drug (i.e., pharmacodynamic sensitivity).3 This phenomenon can be the result of overdose or drug interactions in such highly sensitive patients in whom usual drug dosages lead to extreme elevations in plasma concentration. The latter most commonly occurs when the culprit drug is eliminated by a single metabolizing pathway. If this pathway is susceptible to inhibition by the administration of a second drug, inhibited by coadministration of other drugs or by genetic factors, marked elevation of drug concentrations and electrophysiological toxicity can result. This was the case with terfenadine4 and cisapride, both of which are eliminated as noncardioactive metabolites by the intestinal and hepatic P-450 system, CYP3A4. Many commonly used drugs are potent CYP3A4 inhibitors, such as erythromycin, ketoconazole, and some calcium channel blockers, notably mibefradil, which has been withdrawn from the market. Of note, grapefruit juice markedly inhibits the activity of intestinal but not hepatic CYP3A4. Although CYP3A4 is the most important drug-metabolizing enzyme expressed in the liver and elsewhere, some drugs use other pathways. Among other members of the CYP superfamily, genetically determined polymorphisms have been described in three: CYP2D6, CYP2C9, and CYP2C19. A subset of the population absolutely lacks catalytic activity, so that plasma concentrations of substrate drugs are much higher in such “poor metabolizer” patients. The activity of the P-450 CYP2D6, for example, is absent in approximately 7% of whites and in persons of African heritage. In a situation in which CYP2D6 is the sole eliminating pathway, patients with this “poor metabolizer” trait can therefore display markedly aberrant drug concentrations. Administration of a drug can unmask a subclinical (forme fruste) monogenic arrhythmia syndrome. Mutations in genes (KCNQ1, KCNH2, and SCN5A) causing congenital forms of the long QT syndrome have been identified in patients with acquired forms of long QT syndrome. The exact contribution and mutation frequency cannot be estimated. Mutations in the relevant genes have only a minor role, and rare variations in genes responsible for congenital long QT syndrome seems to be frequent in drug-induced long QT syndrome.5 A subclinical form of the Brugada syndrome can similarly predispose affected patients to the development of drug-induced forms of the Brugada syndrome (www.brugadadrugs.org),6 as a consequence of polymorphisms or mutations in SCN5A or other genes. In these monogenic syndromes, penetrance of the electrocardiographic phenotype can be highly variable, an effect often attributed to unidentified modifier genes. In addition, genetic polymorphisms can increase the risk for drug-induced sudden cardiac death. For example, approximately 10% of African Americans carry a variant in their sodium channel gene that results in a tyrosine (Y) rather than a serine (S) at position 1103 of the protein.7 Study of this variant protein in vitro indicates that it confers changes consistent with the sodium channel–linked variant of long QT syndrome. Similarly, the polymorphisms resulting in T8A and Q9E in the KCNE2 gene have been associated with unusual in vitro characteristics and a higher incidence of proarrhythmia during drug therapy. Drug effects are the most common cause of acquired long QT syndrome. Although syncope occurring soon after the initiation of quinidine therapy was recognized as early as the 1920s, it was not until the advent of continuous electrocardiographic monitoring that “quinidine syncope” was recognized to be due to polymorphic ventricular tachycardia. In recent years, it has become apparent that in addition to antiarrhythmic drugs, a large spectrum of noncardiac drugs (Table 101-1) can cause QT prolongation and torsades de pointes. Dessertenne7a used the term torsades de pointes initially to describe this characteristic undulating pattern of tachycardia in 1966. It is characterized by a polymorphic, usually unsustained, pause-dependent ventricular tachycardia that almost always only develops in association with an excessively prolonged QT interval. It can be associated with syncope or degenerate into a sustained ventricular tachycardia or ventricular fibrillation. An ECG recorded just before or after termination of a polymorphic ventricular tachycardia helps to distinguish torsades from other polymorphic ventricular tachycardias mainly occurring in patients with structural heart disease. Features that are diagnostic for torsades de pointes include a prolonged QT interval, the presence of U waves before the onset or after the termination of the arrhythmia, relatively long coupling intervals, and a typical initiating sequence. In drug-induced long QT syndrome, short-long-short cycle length changes constitute the typical pattern of initiation of torsades de pointes and can be regarded as a warning sign of an impending episode of torsades. Premature ventricular beats often are found to arise from exaggerated T/U waves after longer intervals, such as after extrasystolic pauses (Figure 101-1). Table 101-1 Drugs That Can Cause QT Prolongation or Torsades de Pointes Data derived from non-randomized studies of the literature. This list is not comprehensive; rather, it gives examples of how drugs may be classified according to the criteria proposed in Box 101-1. The reader is recommended to review the literature on any of these agents or drugs of the same drug group before making decisions that relate to their administration. Please see also www.torsades.org. Figure 101-1 Monitor strip showing long-short ventricular cycle, pause-dependent QT prolongation with abnormal and gradually augmented T/U waves (asterisk) leading to a self-terminating episode of torsades de pointes demonstrating the typical “twisting” morphology. Ventricular premature beats are followed by a postextrasystolic pause. The initial beat starts from the peak of the U wave. This patient was taking a combination of clarithromycin and ketoconazole. Torsades de pointes is a major safety concern with drugs that are submitted for regulatory approval. Of concern is that the proarrhythmic risk for many of the drugs that can prolong repolarization, thereby causing torsades de pointes, is not detected during the developmental phase and is recognized only after such drugs have been on the market for many years. In larger series of cases of torsades de pointes, the death rate was up to 16%. In a survey in the United Kingdom and Italy, noncardiovascular drugs that have proarrhythmic potential (i.e., an official warning regarding risk for QT prolongation or torsades de pointes, or with published data on QT prolongation, ventricular tachycardia, or class III effect) represented 3% and 2% of total prescriptions in both countries, respectively.8 The growing awareness of drug-induced long QT syndrome over the past several years has resulted in a marked increase in the number of spontaneous reports. However, the spontaneous reporting most likely underestimates the true incidence of serious adverse reactions, so that the true incidence of drug-induced torsades de pointes is still uncertain. Although torsades de pointes preferentially occurs shortly after initiation of therapy, it can also develop during long-term treatment. The late occurrence has been linked to changes in dose, reinitiation of the drug after short discontinuation, and transient electrolyte disorders such as hypokalemia or hypomagnesemia. The incidence of torsades de pointes associated with sotalol has been estimated to range between 1.8% and almost 5%. The torsadogenic potential of drugs is extremely variable, even within the same class of drugs.9 Some drugs can provoke torsades de pointes only in the setting of overdose or with concomitant administration of other QT-prolonging drugs, or in the presence of risk factors such as hypokalemia, whereas others can induce torsades de pointes when used alone, even at therapeutic levels, and in the absence of additional risk factors. In an attempt to provide a systematic classification of QT-prolonging drugs, a classification of the torsadogenic potency of proarrhythmic drugs (Box 101-1 and Box 101-2) has been proposed.10
Drug-Induced Ventricular Tachycardia
Overview
Pharmacokinetic Risk Factors
Genetic Predisposition
Drug-Induced Long QT Syndrome
Drug-Induced Ventricular Tachycardia



