Drug-Induced Pulmonary Disease Due to Nonchemotherapeutic Agents
INTRODUCTION
Drugs have long been recognized as having the potential to cause pulmonary injury. The precise incidence of drug-induced lung disease is difficult to ascertain, because the signs and symptoms of disease are shared by many other pulmonary conditions and diseases. An analysis of a database of approximately 9 million patients from the United Kingdom describes an incidence density of 0.7 per 100,0000 patient years for interstitial disease related to drugs over a 12-year period (1997–2008).1 These data reflect only a portion of the impact of drug-induced respiratory disease, because the alveoli, upper and lower airways, pleura, pulmonary vasculature, muscles of respiration, and the central nervous system governing respiratory control are all susceptible to injury from ingested, inhaled, and parenterally administered agents. As the categories and varieties of therapeutic drugs continue to increase, clinicians will encounter disease from new culprit drugs in addition to well-established drug reactions. Web-based data repositories, such as www.pneumotox.com, can serve as useful tools for the clinician, as they provide frequent updates based on the emerging literature on drug toxicities.
As the clinician-scientist explores the literature on drug-induced lung injury, it is critical to recognize that not all associations between drug use and respiratory dysfunction imply a definitive causal link between a specific drug and the injury pattern described. The literature needs to be cautiously interpreted before concluding that the reported associations are actually due to the implicated drug rather than a confluence of clinical conditions. This chapter addresses a broad array of drug classes implicated in pulmonary toxicity. Chemotherapeutic agents are discussed separately.
GENERAL PRINCIPLES OF DRUG-INDUCED LUNG DISEASE
The lung has an enormous surface area on which blood-borne substances (therapeutic medications, nutritional supplements, illicit drugs, or toxins) actively interact with lung tissue. Drug-related pulmonary toxicity, however, is a rare event. Reactions typically occur in a small minority of individuals exposed to a given agent. In most cases, lung injury appears to be an idiosyncratic event and cannot be predicted by dose, latency from drug initiation, duration of exposure, or pharmacologic characteristics of the drug. Often, there is no characteristic clinical presentation or pathognomonic histologic pattern of injury associated with a given drug, although certain histologic patterns of lung injury may occur more frequently than others. Thus, establishment of a diagnosis of lung toxicity is frequently a diagnosis of exclusion (see Table 66-1).
It may be inferred from the idiosyncratic nature of toxicity that there are host-specific risk factors that influence development of pulmonary toxicity. The factors influencing individual susceptibility may be: (1) genetically determined, (2) due to concurrent exposures to medications or environmental factors, (3) related to the individual’s comorbid disease, or (4) a function of a combination of these factors.
Genetic predisposition to drug-induced injury is not fully characterized. While drug metabolism occurs primarily in the liver, through the action of the cytochrome P450 family of enzymes, the lung also is an active site of drug biotransformation, with cytochrome P450 enzyme levels estimated as 10% to 15% of those in the liver. In addition, the existence of lung-specific cytochrome P450 isoenzymes implies lung-specific metabolism of drugs.2 Host-specific enzyme polymorphisms affecting drug metabolism may confer increased risk of toxicity on certain individuals.3,4
MECHANISMS OF PULMONARY INJURY
Mechanisms of lung injury include: (1) oxidant injury, (2) immunological and inflammatory cell–mediated injury (including immune complex–mediated injury), (3) interference with matrix formation, (4) abnormal protease/antiprotease balance, and (5) interference with lipid metabolism.
Mechanisms of lung toxicity are less well characterized than are other organ toxicities. The role of drug-induced oxidant injury is well established for nitrofurantoin, and it may be the mechanism of injury of other drugs, as well. Biotransformation of these drugs results in generation of reactive oxygen species, including hydrogen peroxide (H2O2), the hydroxyl radical (•HO), and superoxide anion (O2–•), which promote lipid peroxidation, glutathione depletion, and, consequently, cellular dysfunction or cell death.2
Immunologically mediated injury is undoubtedly important as well. The observation that drug metabolite–protein adducts can act as immunogens, resulting in hypersensitivity reactions or other immunologically mediated tissue injury likely applies to the lung, as well as to other organs.5,6 Lymphocytic or neutrophilic alveolitis and inflammatory cell interstitial infiltrates are present in many cases of drug-induced lung injury, and the elaboration of chemokines and proteases by these cells may lead to cellular injury. Complement-mediated injury has been implicated for drugs causing noncardiogenic pulmonary edema (ARDS), particularly opiates and β-agonists.
Amphiphilic compounds, such as amiodarone, quinidine, and some β-blockers are passively sequestered in the lung within macrophages and type II alveolar cells. The role of disruption of phospholipid metabolism as a consequence of this sequestration has been well established for amiodarone-mediated lung injury, as discussed below.
 IMPACT OF CONCURRENT EXPOSURES OR CONDITIONS
IMPACT OF CONCURRENT EXPOSURES OR CONDITIONS
The harmful effect of concurrent oxygen administration in association with the chemotherapeutic agent, bleomycin, has been well established. It has been suggested that the apparent increased risk of amiodarone toxicity in patients who have had thoracic surgery (see below) may be a result of the impact of intraoperative high oxygen tensions.7 However, high oxygen tension does not substantially increase the risk of toxicity for most nonchemotherapeutic agents. It is plausible that the additive effect of oxidant injury may be more relevant for some individuals than others. There are evolving data that other exogenous factors, such as cigarette smoke, may influence lung injury through induction of cytochrome P450 enzymes.
DIAGNOSTIC APPROACH TO THE PATIENT WITH SUSPECTED DRUG-INDUCED LUNG DISEASE
Confirmation of suspected drug-induced pulmonary toxicity is often a diagnostic challenge. No definitive criteria exist by which to establish the diagnosis of drug-induced disease, but the diagnosis may be inferred if: (1) there is a history of drug exposure; (2) clinical, radiologic, and histopathology are consistent with previously reported toxicity; (3) alternate diagnoses are excluded (e.g., infection, systemic or idiopathic diseases); and (4) findings regress with drug discontinuation and/or treatment. Recrudescence of the lung injury following rechallenge with the culprit drug is further confirmation of drug-induced disease, but rechallenge is not recommended due to the substantial risk of morbidity and/or mortality.
Recognition of a patient’s risk of drug-induced injury is the first step in the diagnostic process. Drug-induced lung disease occurs with prescribed drugs, over-the-counter drugs, herbal or alternative medicine preparations (many of which contain a variety of substances that could be implicated as culprit agents), and illicit drugs. Patients may be reluctant to offer accurate information about their use of alternative medicines or supplements, and the clinician must skillfully elicit the history.
The challenge of diagnosis is further compounded by the fact that the latency from the onset of drug use to development of a toxic reaction can be highly variable, such that the temporal relationship between the pulmonary findings and the culprit drug is not readily apparent. Furthermore, many drugs (e.g., amiodarone and nitrofurantoin) can cause acute, subacute, or chronic pulmonary toxicity. Moreover, new drugs will continue to come on the market, some of which will inevitably cause lung disorders. Often the potential for drugs to cause toxic reactions will only be recognized once the drug has been in use for a sufficient length of time to allow a low frequency event, such as drug-induced toxicity, to be recognized.
In many cases, drug reactions are idiopathic, rather than dose-dependent reactions, and are unpredictable in that they are unrelated to the drug’s intended pharmacologic properties. Exceptions to these observations include (1) amiodarone, for which there is an increased risk of toxicity with higher daily maintenance dosages, and for which the associated histopathology is related to amiodarone’s pharmacologic properties; and (2) heroin, methadone, aspirin, propoxyphene, ethchlorvynol, and colchicine that cause pulmonary toxicity only in the setting of overdose.
A diagnosis of drug-induced lung toxicity is often difficult to establish, because patients with this condition typically come to medical attention with nonspecific symptoms, radiologic findings, and laboratory data. The clinical presentation of drug-induced lung disease may be similar to that of other disorders, including infection, hypersensitivity pneumonitis due to environmental antigens, eosinophilic lung disease, systemic rheumatologic or collagen vascular disease, and idiopathic interstitial pneumonias. High-resolution computed tomography (HRCT) has utility in characterizing the lung injury, but it does not confidently predict the histologic pattern of drug-induced injury.8,9 Even in cases in which lung tissue is obtained by biopsy, the histopathologic features may not fully distinguish drug-induced disease from the disorders or conditions mentioned earlier.
An additional consideration is that the underlying disease for which a drug is given may produce pulmonary findings similar to drug-induced lung disease. For example, rheumatoid arthritis may cause pulmonary infiltrates with similar radiographic appearance and histology to toxic reactions induced by the methotrexate, gold, or penicillamine used to treat the rheumatoid arthritis.
Once the clinician has established that a patient has (1) a recognized, identifiable risk of drug-induced lung toxicity, and (2) history, physical examination, laboratory data, and radiographic findings compatible with known patterns of lung injury, then further cytopathologic or histologic evidence may be warranted to further establish the diagnosis. The technique of bronchoalveolar lavage (BAL) is used in the diagnostic evaluation of patients with interstitial lung diseases (ILDs). Analysis of BAL fluid is most useful when it is used in conjunction with a comprehensive clinical history and HRCT.10 BAL cytologic analysis and culture can be particularly useful to exclude typical or atypical bacterial, fungal, viral, or parasitic infections in patients with suspected drug-induced lung injury. BAL may be helpful in diagnosing diffuse lung malignancies, such as lymphangitic spread of tumor or diffuse pulmonary lymphoma.11 BAL can also provide evidence of alveolar hemorrhage, whether bland or vasculitic in origin. The grossly cloudy BAL fluid that settles with gravity is consistent with pulmonary alveolar proteinosis, which is a reported pattern of injury associated with disease-modifying, antirheumatoid arthritis drugs.12
The cellular differential of the BAL fluid may help narrow the diagnosis in suspected drug-induced pulmonary disease. The most common BAL cellular profile in drug-induced disease is lymphocytosis; a cell differential of ≥25% lymphocytes should earn drug-induced ILD a place on the differential diagnosis of any presentation of ILD. Recent guidelines for the use of BAL do not recommend routine measurement of lymphocyte subsets on all patients with ILD. However, if a lymphocyte alveolitis is suspected or confirmed, the subset measurement may be justified.10 The T suppressor (CD8+) lymphocyte subset is commonly reported in drug-induced lung disease, as seen in hypersensitivity pneumonitis. BAL T helper (CD4+) predominance has been associated with immunomodulatory drug injury, including methotrexate, sirolimus, and temsirolimus.13,14 Other cellular profiles occur in drug-induced disease: Eosinophils and neutrophils are variably present.15–17 A percentage of eosinophils ≥25% is consistent with acute or chronic eosinophilic pneumonia, which may be seen as a result of drug injury.
Lung tissue obtained bronchoscopically or surgically may help characterize the injury pattern and may support the diagnosis of drug-induced injury. However, lung biopsy is not essential to establish the diagnosis. The histologic patterns seen in drug-induced injury, whether usual or nonspecific interstitial pneumonia (NSIP), organizing pneumonia with or without bronchiolitis obliterans, eosinophilic pneumonia, or granulomatous inflammation are histologically indistinguishable from other etiologies of these lung injury patterns.
Adjunctive testing for serum markers has been studied in the context of drug-induced lung injury. Serum markers, including surfactant proteins (SP-A, SP-D), KL-6, and ADAM8 have been associated with drug-induced injury but have not entered into the diagnostic armamentarium for most routine clinical evaluations.18–21 Similarly, drug-specific lymphocyte stimulation testing and lymphocyte migration inhibition testing have been shown to be useful in the diagnosis of drug-induced lung disease by some investigators, but have not been found to be sufficiently and clinically helpful to enter mainstream clinical practice.22–25
CLINICAL AND HISTOPATHOLOGIC PATTERNS OF INJURY
The entire respiratory system – upper and lower airway, pleura, lung parenchyma, pulmonary vasculature, muscles of respiration, and the central nervous system governing respiratory control – is susceptible to the adverse effects of drugs. Of these areas, injury to the parenchyma is the most important cause of morbidity and mortality.
Respiratory disease may occur as the sole consequence of drug toxicity, or it may be one manifestation of a systemic drug-induced syndrome. For example, systemic hypersensitivity syndromes, such as drug rash with eosinophilia and systemic symptoms (DRESS), may be induced by drugs, particularly the aromatic anticonvulsants.26 Drug-induced systemic lupus erythematosus (SLE) may occur, with or without pulmonary involvement, from exposure to β-blockers, amiodarone, angiotensin-converting enzyme inhibitors (ACEIs), hydralazine, procainamide, isoniazid, methyldopa, minocycline, and tetracycline, among others. In addition, drugs (e.g., phenytoin, hydralazine, propylthiouracil, D-penicillamine, and cocaine, among others) can cause the clinical picture of a pulmonary–renal syndrome, with evidence for pulmonary and renal vasculitis with renal failure.
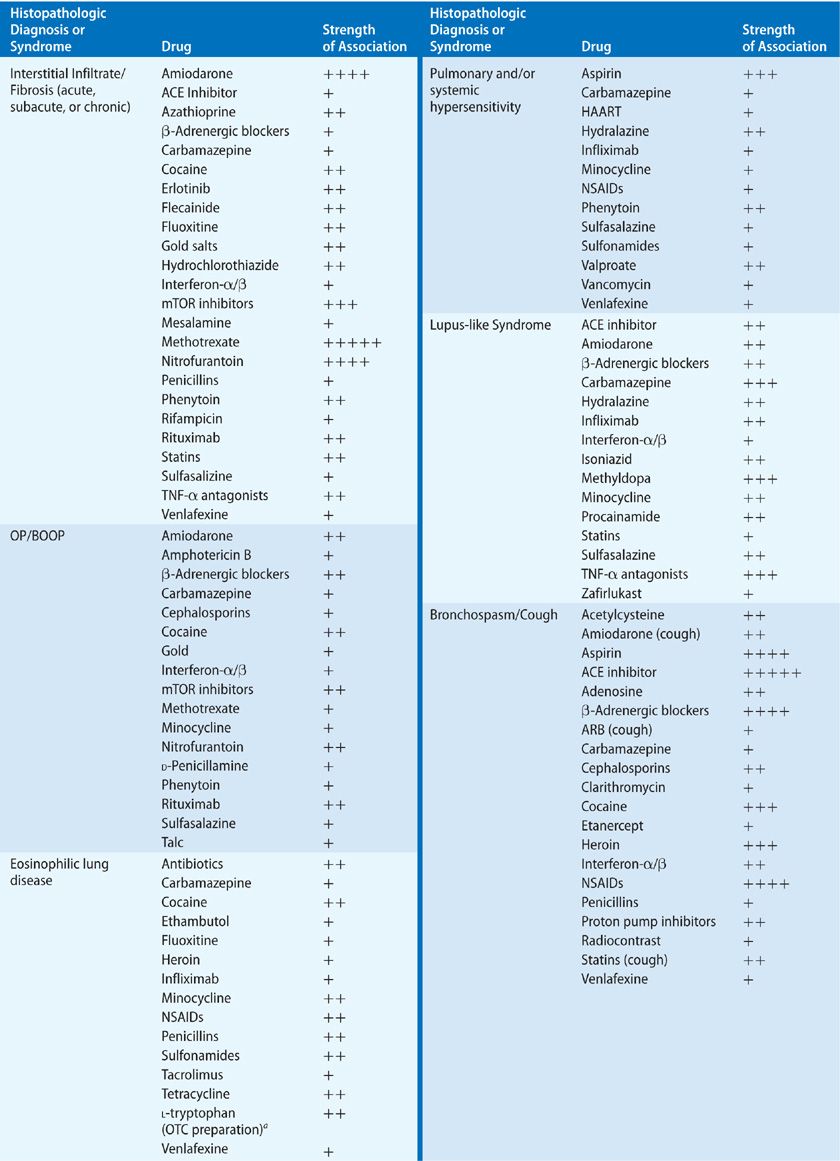
Parenchymal injury may manifest itself in the interstitial, alveolar, and/or vascular compartments. Of the processes affecting the lung parenchyma, interstitial involvement is the most common. The major histopathologic forms of ILD – cellular and fibrotic NSIP and usual interstitial pneumonia (UIP) – have been reported to occur as an adverse effect of drugs. Unfortunately for diagnostic clarity, among the many case reports citing the presence of ILD, many have no tissue confirmation of the precise lung histology, and older case reports were published before the current guidelines for classification of ILDs were established. However, it seems likely that much of the previously reported drug-induced ILD would now be classified as either cellular or fibrotic NSIP. Virtually all histopathologic types of ILD have been reported to occur in association with drugs, including organizing pneumonia (with and without obliterative bronchiolitis), usual interstitial pneumonitis, eosinophilic pneumonia, desquamative interstitial pneumonitis, and hypersensitivity pneumonitis. It is important to recognize that few drugs have been reported to cause a single histopathologic pattern of parenchymal injury, and in the cases of many drugs, several patterns of injury may occur (Table 66-2).
 NONSPECIFIC AND USUAL INTERSTITIAL PNEUMONIAS
NONSPECIFIC AND USUAL INTERSTITIAL PNEUMONIAS
The radiographic and histologic definitions of NSIP and UIP have been defined in the American Thoracic Society/European Respiratory Society (ATS/ERS) international, multidisciplinary consensus statements of 2002 and 2013.27,28 Whether drug-induced, idiopathic, or due to systemic disease, the radiographic and histologic features are largely indistinguishable, and it is the clinical scenario that is critical to the differential diagnosis. Older literature on drug-induced lung disease does not necessarily conform to recent consensus definitions of NSIP, UIP, or otherwise unclassified interstitial pneumonias, making older case series somewhat difficult to interpret. Culprit drugs leading to these interstitial pneumonias include antimicrobials, antirheumatics, tumor necrosis factor (TNF) antagonsists, β-adrenergic antagonists, antiarrhythmics, anti-inflammatory drugs, antipyretics, and immunosupressants (see Table 66-2).
 BRONCHIOLITIS OBLITERANS AND ORGANIZING PNEUMONIA
BRONCHIOLITIS OBLITERANS AND ORGANIZING PNEUMONIA
Organizing pneumonia (OP) with or without histopathologic evidence of obliterative bronchiolitis is a frequently reported pulmonary reaction to medications. The histologic pattern seen in drug-induced parenchymal injury is indistinguishable from cryptogenic organizing pneumonia (COP) or other causes of bronchiolitis obliterans organizing pneumonia (BOOP). The histology consists of plugs of loose connective tissue, or granulation tissue that fills respiratory bronchioles, alveolar ducts, and alveolar spaces. This may be accompanied by mild interstitial inflammation with preserved architecture. Radiographic findings of BOOP are typically bilateral, patchy areas of consolidation that are often subpleural, with a tendency to lower lobe predominance or distribution along the bronchovascular bundle.
Many of the drugs that have been reported to cause BOOP are commonly used medications. Among the implicated antimicrobials are cephalosporins, minocycline, nitrofurantoin, amphotericin B, and interferons. One of the most utilized antiarrhythmic agents, amiodarone, is known to cause BOOP, as are the anticonvulsants, carbamazepine and phenytoin, and the anti-inflammatory agents, gold, penicillamine, and sulfasalazine. In patients treated for rheumatoid arthritis (RA) with gold or penicillamine, it is important to distinguish between drug-induced OP and infiltrates reflecting a pulmonary manifestation of the underlying RA itself. A variety of other agents reported to cause obliterative bronchiolitis are listed in Table 66-2.
The clinical presentation of drug-associated OP or BOOP is similar to that of the idiopathic disease, which is now referred to as COP. Symptoms include shortness of breath, nonproductive cough, and, in some cases, low-grade fever and/or pleuritic chest pain. The chest radiograph typically shows bilateral patchy infiltrates that may be migratory over serial radiographs, with interval normal chest radiographs despite continuous drug exposure. As with other interstitial lung disease, the utility of BAL is primarily to exclude an infectious etiology of the infiltrates. There is no specific BAL cellular profile characteristic of OP or BOOP. Lung biopsy reveals characteristic histopathology, identical to that of COP. Patients with drug-induced OP or BOOP may have spontaneous resolution of disease when the offending drug is discontinued, but oral corticosteroids may be used to accelerate disease resolution if the patient is symptomatic.
 EOSINOPHILIC LUNG DISEASE
EOSINOPHILIC LUNG DISEASE
Drug-induced eosinophilic lung disease may mimic other eosinophilic pulmonary syndromes, including simple eosinophilic pneumonitis (Loeffler’s syndrome), chronic eosinophilic pneumonia, acute eosinophilic pneumonia, pulmonary infiltrates with peripheral eosinophilia (PIE), and Churg–Strauss syndrome (see Chapter 71). The differential diagnosis of eosinophilic lung disease includes drug-induced injury and a search for the culprit drug is an integral part of the diagnostic evaluation. Although the clinical presentation of drug-induced eosinophilic pneumonia may be identical to idiopathic conditions, several distinctions can be made. In idiopathic eosinophilic pneumonia, symptoms affect the lung exclusively, while in drug-induced eosinophilic pneumonia, respiratory symptoms may be accompanied by systemic symptoms, such as rash and fever. Marked peripheral blood eosinophilia (>1000 cells/mL) suggests drug-induced pneumonitis, rather than acute idiopathic eosinophilic pneumonia, in which the eosinophilia tends to be more modestly elevated or normal.
The diagnosis of drug-induced eosinophilic pneumonia is supported by peripheral blood and/or pulmonary eosinophilia in a setting of exposure to a suspect drug. The diagnosis may be established when other eosinophilic lung diseases are excluded. When evaluating a patient with pulmonary eosinophilia, it is particularly important to exclude infectious causes of eosinophilia so as to avoid promoting progressive infection and/or death by use of corticosteroid treatment for presumptive drug-induced eosinophilic pneumonia. Tropical pulmonary eosinophilia caused by filarial infection should be suspected if the patient has a consistent travel history. Schistosoma and Paragonimus westermani are other potential pathogens to be excluded. Strongyloides, Ascaris, and Toxocara are indigenous to the United States and are known to cause pulmonary infiltrates and peripheral blood eosinophilia. Missing the diagnosis of a fungal infection can be lethal. Aspergillus is a ubiquitous fungus that can be difficult to diagnose as a pulmonary pathogen. BAL eosinophilia may be present without definitive evidence of invasive fungal infection, which may require a tissue biopsy for diagnosis. Coccidioides immitis is endemic in the southwestern United States, and infection can result in peripheral blood eosinophilia and pulmonary infiltrates. Serologic testing for antibodies to Coccidioides and sputum or BAL cultures are useful studies to exclude coccidioidomycosis.
Specific testing for drug reactions, such as drug-specific lymphocyte transformation tests, has been used to implicate a culprit drug. However, the clinical utility of such tests is uncertain, especially because a negative assay does not exclude the diagnosis.
Successful management of drug-induced eosinophilic lung disease is achieved by identification and discontinuation of the inciting drug. Typically, resolution of symptoms in drug-induced eosinophilic lung disease occurs with discontinuation of the culprit drug, frequently without the need for corticosteroids. In contrast, idiopathic chronic eosinophilic pneumonia may require months of treatment with corticosteroids, and relapse may occur as steroids are tapered. Relapse of the disease as steroids are tapered is rare in drug-induced eosinophilic disease; recrudescence of the lung infiltrates suggests an alternate diagnosis.
 HYPERSENSITIVITY SYNDROMES
HYPERSENSITIVITY SYNDROMES
Systemic hypersensitivity syndromes with granulomatous inflammation of the lung may be caused by a number of drugs, most commonly the aromatic anticonvulsants, phenytoin, and carbamazepine, as well as nonsteroidal anti-inflammatory drugs (NSAIDs), minocycline, and sulfonamides, among others. DRESS has been reported primarily with the anticonvulsants and, in some cases, is accompanied by pulmonary histopathology consistent with BOOP, ILD, or granulomatous inflammation.
Not all cases of pulmonary hypersensitivity are accompanied by rash or other systemic symptoms. The clinical presentation of drug-related pulmonary hypersensitivity typically consists of acute onset of dyspnea, cough, and fever. The radiographic pattern is one of diffuse reticular or peripheral alveolar infiltrates, sometimes accompanied by pleural effusion. In most cases, drug discontinuation results in disease resolution. A minority of individuals (10%) have persistent radiographic abnormalities after several months, and rarely, progressive disease may occur despite drug withdrawal. In these cases, a trial of corticosteroids is reasonable if the diagnosis has been well established.
 ALVEOLAR INJURY: NONCARDIOGENIC PULMONARY EDEMA AND ACUTE RESPIRATORY DISTRESS SYNDROME
ALVEOLAR INJURY: NONCARDIOGENIC PULMONARY EDEMA AND ACUTE RESPIRATORY DISTRESS SYNDROME
Alveolar disease occurs in the form of noncardiogenic pulmonary edema, diffuse alveolar damage (DAD) or acute respiratory distress syndrome (ARDS), and both bland and vasculitic diffuse alveolar hemorrhage (DAH). (Vasculitis due to drugs is discussed in the separate section below.)
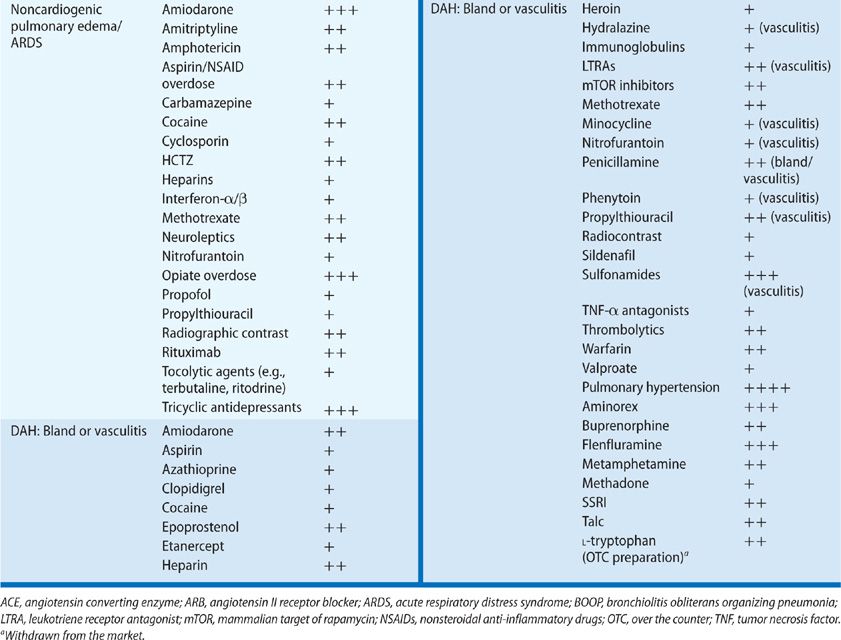
Noncardiogenic pulmonary edema may be precipitated by numerous drugs, including aspirin, opiates, calcium channel blockers, some diuretics (e.g., hydrochlorothiazide and acetazolamide), intravenous and inhaled pulmonary vasodilators (e.g., epoprostenol and nitric oxide), methotrexate, TNF-alpha radiographic contrast media, tocolytics, and oxytocin, among others.29 The onset of drug-induced pulmonary edema typically occurs in close temporal proximity to ingestion of the culprit drug and does not have a variable latency of onset.
A variety of mechanisms are implicated in the development of pulmonary edema. Increased pulmonary capillary permeability is the likely cause of aspirin-induced, interleukin-2–induced, and ethchlorvynol-induced edema. Volume expansion and sodium retention likely cause the pulmonary edema associated with the use of β2-sympathomimetic tocolytic agents. Opioid-induced edema is mediated by neurogenic reflex and deposition of immunoglobulin and complement in the lung.30 Although the mechanisms of pulmonary edema may vary, frequently the resolution of symptoms is prompt within days of drug discontinuation.
Pulmonary edema is common in salicylate intoxication, and the risk is correlated with the degree of intoxication. A prevalence of alveolar edema as high as 20% to 30% has been reported for aspirin-intoxicated adults. An increased risk of edema is associated with chronic aspirin ingestion, older age, positive smoking history, neurologic disease, and proteinuria.31,32
In many cases reported as drug-induced ARDS, this condition is considered as a clinical syndrome of lung injury, rather than a specific histopathologic pattern of disease. If we examine the more specific histopathology of DAD, many cases of drug-induced injury have been reported to have these histologic characteristics and diffuse ground-glass attenuation on CT imaging of the lung. The more commonly implicated drugs associated with DAD are amiodarone, methotrexate, rituximab, and many chemotherapeutic agents.
 DIFFUSE ALVEOLAR HEMORRHAGE, VASCULITIS, AND PULMONARY–RENAL SYNDROMES
DIFFUSE ALVEOLAR HEMORRHAGE, VASCULITIS, AND PULMONARY–RENAL SYNDROMES
Drug-induced DAH is infrequently reported in the literature on drug-induced pulmonary disease. DAH should be suspected in patients with unexplained ground-glass infiltrates or consolidation, accompanied by anemia. Hemoptysis is not required for the diagnosis, and may be absent in as many as one-third of patients with DAH.
The classification of DAH is based on histopathologic findings and includes (1) capillaritis, (2) bland hemorrhage due to drug-induced coagulopathies, and (3) DAD, the histopathologic finding in ARDS.
Pulmonary capillaritis has been described as an adverse effect of many drugs, but strong associations with its development are reported for relatively few drugs. One of the best-characterized toxicities presenting as DAH is that from propylthiouracil.33 The DAH is a manifestation of a systemic vasculitic syndrome characterized by leukocytoclastic vasculitis, glomerulonephritis, and pulmonary capillaritis. Use of propylthiouracil induces antineutrophil cytoplasmic antibodies (ANCAs) in as many as 46% of patients, although only a minority of these develop pulmonary toxicity; pathogenesis may be mediated by antimyeloperoxidase antibodies.34,35 Cases of pulmonary or systemic vasculitis also have been reported for diphenylhydantoin, hydralazine, nitrofurantoin, and leukotriene receptor antagonists. In the case of the last group of drugs noted, there has been considerable discussion as to whether the vasculitis is a toxic effect of the leukotriene antagonists or whether withdrawal of oral corticosteroids leads to identification of pre-existing Churg–Strauss granulomatous vasculitis. At least some reported cases of Churg–Strauss syndrome are unrelated to steroid withdrawal and appear to represent a rare complication of leukotriene antagonists.
Bland hemorrhage without capillaritis can occur (1) in the setting of therapeutic and supratherapeutic anticoagulation, (2) with the use of inhibitors of platelet aggregation (e.g., clopidigrel), and platelet glycoprotein IIa/IIIb inhibitor therapy, and (3) as a complication of thrombolysis. Drug combinations may increase the risk of pulmonary hemorrhage. For example, when therapeutic anticoagulation is combined with epoprostenol for management of pulmonary hypertension, the risk of hemorrhage is compounded.36 Bland DAH related to D-penicillamine, amiodarone, and cocaine is discussed in the sections on individual drugs in later sections.
Drug-induced DAD may lead to bland DAH and has been reported for amiodarone, nitrofurantoin, minocycline, methotrexate, gold, cocaine, and chemotherapeutic agents.
 COUGH AND ANGIOEDEMA
COUGH AND ANGIOEDEMA
Cough induced by ACEIs is well characterized in several case series.36–40 The overall reported incidence of cough ranges from 5% to 25%. Women are more at risk of developing cough than are men.37,39,41 The latency from initiation of use to the onset of cough is variable, ranging from 1 week to 15 months. A prior diagnosis of asthma has not been found to be a significant risk factor for the development of ACEI-associated cough. While some series have reported increased nonspecific bronchial hyperresponsiveness among subjects with cough, others have not. Symptoms typically resolve promptly after a week to 1 month after discontinuation of the drug, but occasionally resolution may take as long as 3 months.41 Angiotensin receptor blockers induce cough in approximately one-third of patients in whom ACEI-induced cough has been diagnosed.42 Less frequent side effects of ACEIs include PIE, SLE, and subacute ILD, but these do not typically appear in conjunction with ACEI-induced cough.
Angioedema to ACEIs is a potentially life-threatening complication of these medications. Edema more commonly affects the face (57%) and less commonly affects the floor or roof of mouth and tongue (26%). In 17%, the oropharynx and glottis are involved.43 Presenting symptoms may range from simple edema of the lips and/or face to respiratory failure requiring ventilatory support. The latency for onset is even wider than that for cough, ranging from 4 weeks to 4 years after onset of ACEI use. In one series, 70% presented within the first month of treatment.44
Numerous case series have reported a higher incidence of angioedema among African Americans, and a higher risk of intubation among those affected in this racial group.43,44 Angioedema formation is likely due to elevation of circulating bradykinin as a result of ACEI-induced inhibition of its degradation, leading to vasodilatation and capillary leak. The effect of impaired bradykinin degradation may be compounded in individuals with genetically based deficiency of other bradykinin metabolizing enzymes. Most experts recommend treatment for angioedema with corticosteroids, H1- and H2-blockers, and, in some case, epinephrine.
 BRONCHOSPASM AND ANAPHYLAXIS
BRONCHOSPASM AND ANAPHYLAXIS
Drug-induced bronchospasm is a relatively common side effect of many categories of medications (see Table 66-2).45 Bronchospasm is mediated by a variety of mechanistic pathways including (1) IgE-mediated anaphylaxis, (2) non–IgE-mediated anaphylactoid reactions, (3) alteration in the cyclooxygenase (COX) and lipoxygenase pathways, and (4) other pharmacologic mechanisms, such as β-blockade.46,47
Brochospasm triggered by drugs is more common in subjects with underlying asthma, atopy, or bronchial hyperreactivity. These reactions are more common in women than in men, and there appears to be familial predisposition to reactions to some drugs.48 Symptoms range from mild chest tightness and dyspnea on exertion to respiratory failure in susceptible individuals.
In the United States, medications are the primary cause of anaphylaxis in adults and antimicrobials have been implicated as the cause of 0.7% to 10% of cases of severe bronchospasm. Penicillin remains the most important cause of anaphylaxis, accounting for up to 75% of cases annually.49,50 Aspirin and NSAIDs are commonly implicated in bronchospasm, in one series accounting for as many as 24% of bronchoconstrictive reactions, ranging from mild to severe reactions.49,51 (Aspirin and NSAIDs are discussed in the sections below.) Other agents more commonly implicated in bronchospasm and anaphylaxis include sulfonamides, omalizumab, halothane, heparins, protamine, insulin, and neuromuscular blocking agents.
 BRONCHIOLITIS
BRONCHIOLITIS
Agents that have been reported to cause small airway inflammation (bronchiolitis) include medications used in the treatment of RA, such as D-penicillamine, gold, and tiopronin. The bronchiolitis in these cases may be fatal.52–55 Interpretation of earlier literature on D-penicillamine and gold-induced bronchiolitis is complicated by the fact that RA itself can cause bronchiolitis.56 Use of these agents has been largely supplanted by disease modifying antirheumatic drugs, immunosuppressants, and biologics, but the implicated agents may still be used in certain cases. Drug-induced bronchiolitis typically causes shortness of breath, with or without wheezing, and cough. Spirometry may not detect small airway disease until obstruction is severe, but high-resolution CT imaging protocols may show centrilobular nodules and/or tree-in-bud opacities, as well as mosaic attenuation of the lung parenchyma, reflective of airtrapping.57
 PULMONARY HYPERTENSION
PULMONARY HYPERTENSION
Pulmonary hypertension (see Chapter 72) is a relatively infrequent complication of drug therapy, but because of the subtlety of the onset of disease, paucity of symptoms until significant vascular compromise has occurred, and potential for eventual vascular collapse and death, it is critical to recognize drug-induced pulmonary hypertension early in its course. Among the drugs known to cause pulmonary hypertension are cocaine, other illicit stimulants, anorexigens, and toxic contaminants of food and nutritional supplements (e.g., tryptophan).58
The association of appetite suppressants with pulmonary hypertension dates to the late 1970s when reports of unexplained “primary” pulmonary hypertension were first published. The “epidemic” of pulmonary hypertension was linked to the use of aminorex fumarate, an amphetamine-derived appetite suppressant that came into use because its potential for addiction and abuse was lower than that of amphetamine. The use of aminorex was associated with a significant rise in the incidence of pulmonary hypertension, primarily among women in Germany, Austria, and Switzerland. Development of disease occurred as early as weeks to months from the onset of use of the drug, with a dose-dependent risk as high as 2 in 100. The mechanism of action on the pulmonary vasculature is through the release of catecholamines, including dopamine. The epidemic subsided as the drug’s use declined.
The aminorex epidemic was followed by the introduction of fenfluramine, a phenylethylamine, like amphetamine and aminorex. Fenfluramine had been shown to be equally effective for weight reduction as an amphetamine, without the potential for abuse.
Satiety is normally accompanied by the release of serotonin, which acts on central serotonin 2C receptors. Fenfluramine and racemic dexfenfluramine mimic normal satiety through competitive inhibition of the serotonin transporter, leading to release of serotonin from intracellular stores. These agents were used primarily in Europe throughout the 1980s. Case reports of users with pulmonary hypertension were published in Britain as early as 1981, but use of these agents persisted through the 1980s.
Two landmark reports supported a causal relationship between pulmonary hypertension and the use of fenfluramine and dexfenfluramine. The first was published in 1993, describing a cohort of young-to-middle–aged users of the anorexigens who developed pulmonary hypertension indistinguishable from idiopathic primary arterial pulmonary hypertension.59 This was followed in 1996 by the findings of the International Primary Pulmonary Hypertension Study Group, which published a case-control series of 95 patients with pulmonary arterial hypertension.60 An odds ratio (OR) of 23.1 for development of disease was found among those who had used an anorexigen for more than 3 months, with an estimated incidence of one to two cases per million users per year. Further support for a causal link is contained in the surveillance study of pulmonary hypertension among anorexigen users in the United States. Fenfluramine was withdrawn from the market in 1995 due to its association with increased risk for pulmonary hypertension, as well as its role in the development of valvular heart disease in users of the fenfluramine–phentermine combination anorexigen, known as “fen-phen.”61
Epidemics of pulmonary hypertension also have been reported due to contaminants in a specific manufacturer’s rapeseed oil in Spain, and from use of an over-the-counter L-tryptophan preparation that resulted in the eosinophilic myalgia syndrome, characterized by a systemic syndrome, which included acute lung injury and pulmonary hypertension.
The reason(s) that some individuals develop drug-related pulmonary hypertension while others do not has not been clearly defined. One putative risk may be polymorphisms of a cytochrome P450 enzyme, CYP 2D6, which is the primary enzyme for metabolism of fenfluramine.62 Other risk factors have yet to be characterized.
TOXICITY ASSOCIATED WITH SELECTED DRUGS
The following drugs have been selected for discussion because (1) they are very commonly used, or (2) have important forms of toxicity associated with their use, or (3) have more recently entered into our pharmacologic armamentarium and, therefore, are likely associated a lower level of awareness of their potential toxicity to the respiratory system.
 CARDIOVASCULAR DRUGS
CARDIOVASCULAR DRUGS
Important examples of cardiovascular agents associated with pulmonary toxicity are highlighted in the sections that follow.
Amiodarone
Amiodarone is an iodinated, benzofuran-derivative antiarrhythmic used for management of life-threatening supraventricular and ventricular arrhythmias. Both amiodarone and its major metabolite, desethylamiodarone, are cationic, amphiphilic compounds with high lipid solubility, causing the drug to accumulate in a variety of tissues. The elimination half-life of amiodarone is 30 to 60 days. However, the concentration of amiodarone in lung tissue can be 100- to 500-fold higher than serum levels and the drug has been found in lung tissue as long as 1 year after discontinuation of therapy. These pharmacokinetic characteristics contribute to the drug’s potential toxicity and impact treatment strategies.
One of amiodarone’s biochemical effects is to impair normal phospholipid catabolism by phospholipases, thereby leading to cellular phospholipidosis. The accumulation of phospholipids in the cell may cause direct cellular injury and secondary tissue inflammation. Evidence supports cellular injury through production of the unstable aryl radical as amiodarone is deiodinated, leading to reactive oxygen species formation and cell death.63
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


