DISORDERS OF LIPOPROTEIN METABOLISM
Daniel J. Rader ![]() Helen H. Hobbs
Helen H. Hobbs
Lipoproteins are complexes of lipids and proteins that are essential for the transport of cholesterol, triglycerides, and fat-soluble vitamins. Previously, lipoprotein disorders were the purview of specialized lipidologists, but the demonstration that lipid-lowering therapy significantly reduces the clinical complications of atherosclerotic cardiovascular disease (ASCVD) has brought the diagnosis and treatment of these disorders into the domain of the internist. The number of individuals who are candidates for lipid-lowering therapy has continued to increase. The development of safe, effective, and well-tolerated pharmacologic agents has greatly expanded the therapeutic armamentarium available to the physician to treat disorders of lipid metabolism. Therefore, the appropriate diagnosis and management of lipoprotein disorders is of critical importance in the practice of medicine. This chapter will review normal lipoprotein physiology, the pathophysiology of primary (inherited) disorders of lipoprotein metabolism, the diseases and environmental factors that cause secondary disorders of lipoprotein metabolism, and the practical approaches to their diagnosis and management.
LIPOPROTEIN METABOLISM
LIPOPROTEIN CLASSIFICATION AND COMPOSITION
Lipoproteins are large macromolecular complexes that transport hydrophobic lipids (primarily triglycerides, cholesterol, and fat-soluble vitamins) through body fluids (plasma, interstitial fluid, and lymph) to and from tissues. Lipoproteins play an essential role in the absorption of dietary cholesterol, long-chain fatty acids, and fat-soluble vitamins; the transport of triglycerides, cholesterol, and fat-soluble vitamins from the liver to peripheral tissues; and the transport of cholesterol from peripheral tissues to the liver.
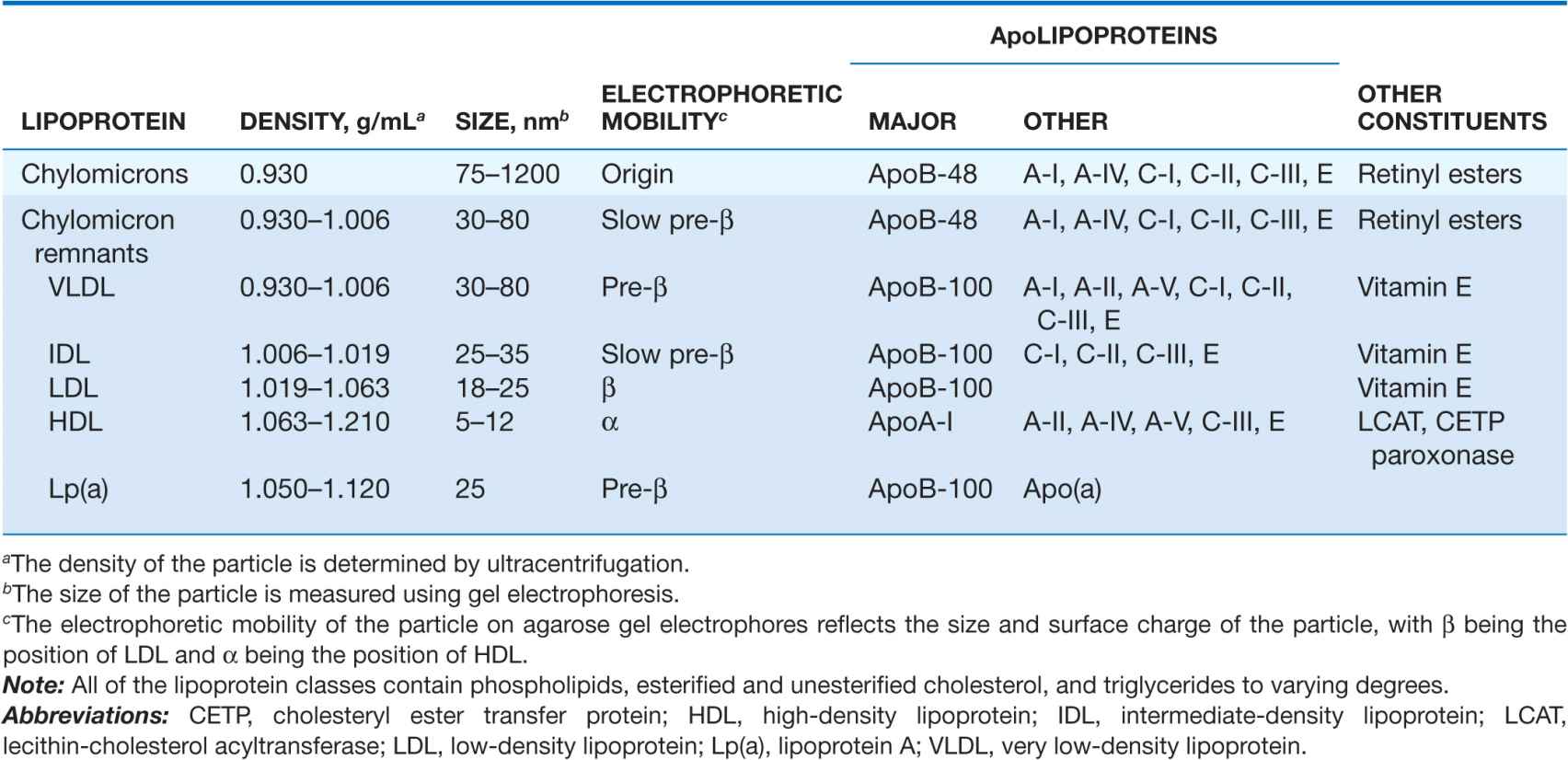
Lipoproteins contain a core of hydrophobic lipids (triglycerides and cholesteryl esters) surrounded by hydrophilic lipids (phospholipids, unesterified cholesterol) and proteins that interact with body fluids. The plasma lipoproteins are divided into five major classes based on their relative density (Fig. 31-1 and Table 31-1): chylomicrons, very low-density lipoproteins (VLDLs), intermediate-density lipoproteins (IDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs). Each lipoprotein class comprises a family of particles that vary slightly in density, size, and protein composition. The density of a lipoprotein is determined by the amount of lipid per particle. HDL is the smallest and most dense lipoprotein, whereas chylomicrons and VLDLs are the largest and least dense lipoprotein particles. Most plasma triglyceride is transported in chylomicrons or VLDLs, and most plasma cholesterol is carried as cholesteryl esters in LDLs and HDLs.
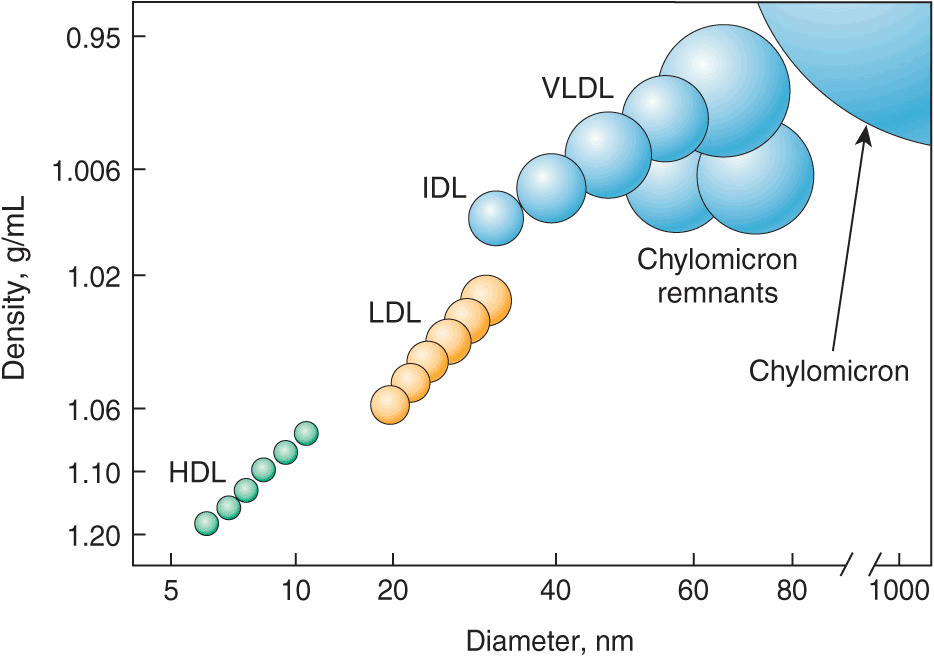
FIGURE 31-1
The density and size distribution of the major classes of lipoprotein particles. Lipoproteins are classified by density and size, which are inversely related. VLDL, very low-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; HDL, high-density lipoprotein.
TABLE 31-1
MAJOR LIPOPROTEIN CLASSES
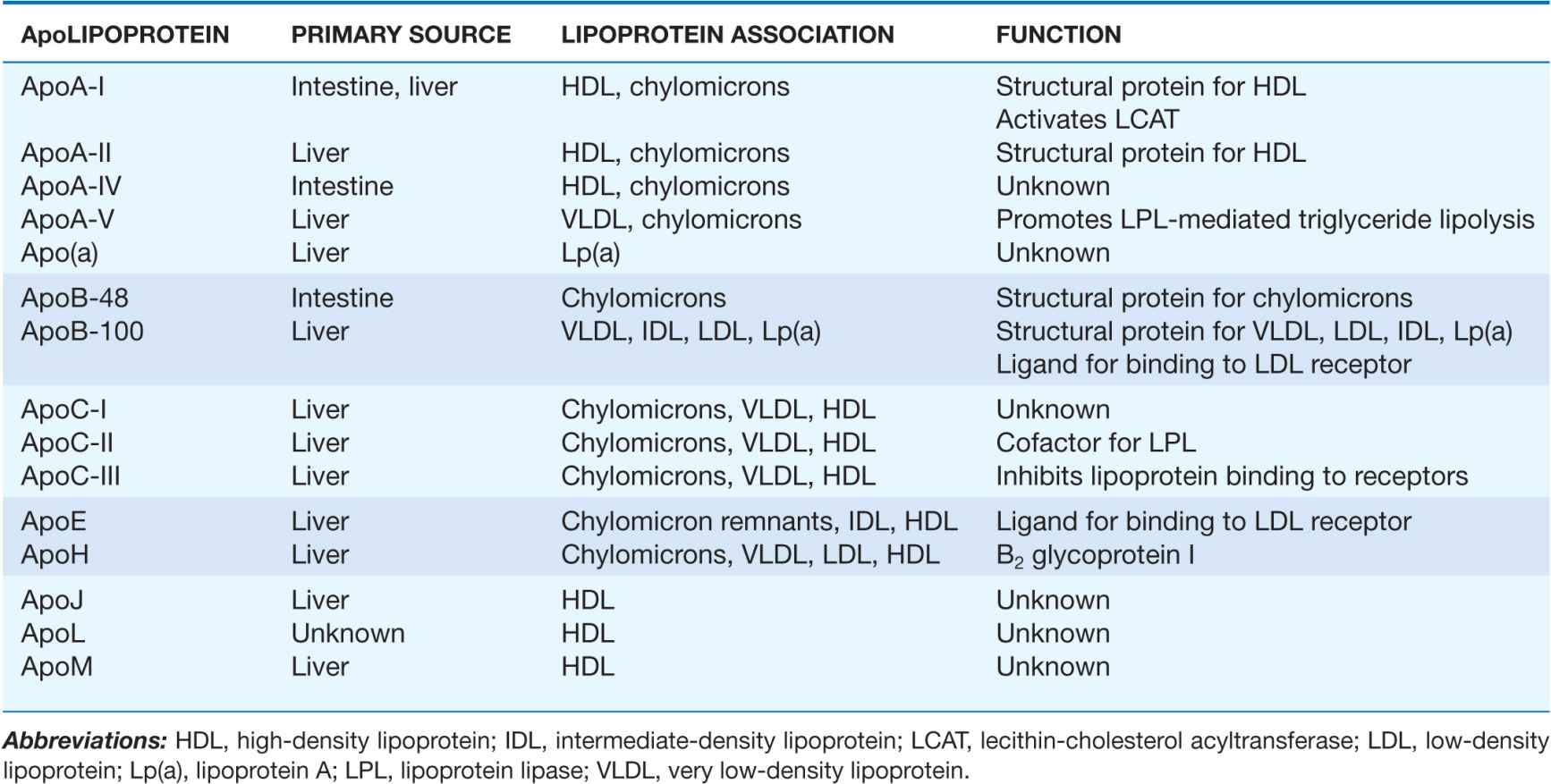
The proteins associated with lipoproteins, called apolipoproteins (Table 31-2), are required for the assembly, structure, and function of lipoproteins. Apolipoproteins activate enzymes important in lipoprotein metabolism and act as ligands for cell surface receptors. ApoA-I, which is synthesized in the liver and intestine, is found on virtually all HDL particles. ApoA-II is the second most abundant HDL apolipoprotein and is on approximately two-thirds of the HDL particles. ApoB is the major structural protein of chylomicrons, VLDLs, IDLs, and LDLs; one molecule of apoB, either apoB-48 (chylomicron) or apoB-100 (VLDL, IDL, or LDL), is present on each lipoprotein particle. The human liver synthesizes apoB-100, and the intestine makes apoB-48, which is derived from the same gene by mRNA editing. ApoE is present in multiple copies on chylomicrons, VLDL, and IDL, and it plays a critical role in the metabolism and clearance of triglyceride-rich particles. Three apolipoproteins of the C-series (apoC-I, apoC-II, and apoC-III) also participate in the metabolism of triglyceride-rich lipoproteins. ApoB is the only major apolipoprotein that does not transfer between lipoprotein particles. Some of the minor apolipoproteins are listed in Table 31-2.
TABLE 31-2
MAJOR ApoLIPOPROTEINS
TRANSPORT OF DIETARY LIPIDS (EXOGENOUS PATHWAY)
The exogenous pathway of lipoprotein metabolism permits efficient transport of dietary lipids (Fig. 31-2). Dietary triglycerides are hydrolyzed by lipases within the intestinal lumen and emulsified with bile acids to form micelles. Dietary cholesterol, fatty acids, and fat-soluble vitamins are absorbed in the proximal small intestine. Cholesterol and retinol are esterified (by the addition of a fatty acid) in the enterocyte to form cholesteryl esters and retinyl esters, respectively. Longer-chain fatty acids (>12 carbons) are incorporated into triglycerides and packaged with apoB-48, cholesteryl esters, retinyl esters, phospholipids, and cholesterol to form chylomicrons. Nascent chylomicrons are secreted into the intestinal lymph and delivered via the thoracic duct directly to the systemic circulation, where they are extensively processed by peripheral tissues before reaching the liver. The particles encounter lipoprotein lipase (LPL), which is anchored to a glycosylphosphatidylinositol-anchored protein, GPIHBP1, that is attached to the endothelial surfaces of capillaries in adipose tissue, heart, and skeletal muscle (Fig. 31-2). The triglycerides of chylomicrons are hydrolyzed by LPL, and free fatty acids are released. ApoC-II, which is transferred to circulating chylomicrons from HDL, acts as a required cofactor for LPL in this reaction. The released free fatty acids are taken up by adjacent myocytes or adipocytes and either oxidized to generate energy or reesterified and stored as triglyceride. Some of the released free fatty acids bind albumin before entering cells and are transported to other tissues, especially the liver. The chylomicron particle progressively shrinks in size as the hydrophobic core is hydrolyzed and the hydrophilic lipids (cholesterol and phospholipids) and apolipoproteins on the particle surface are transferred to HDL, creating chylomicron remnants. Chylomicron remnants are rapidly removed from the circulation by the liver through a process that requires apoE as a ligand for receptors in the liver. Consequently, few, if any, chylomicrons or chylomicron remnants are present in the blood after a 12-h fast, except in patients with disorders of chylomicron metabolism.
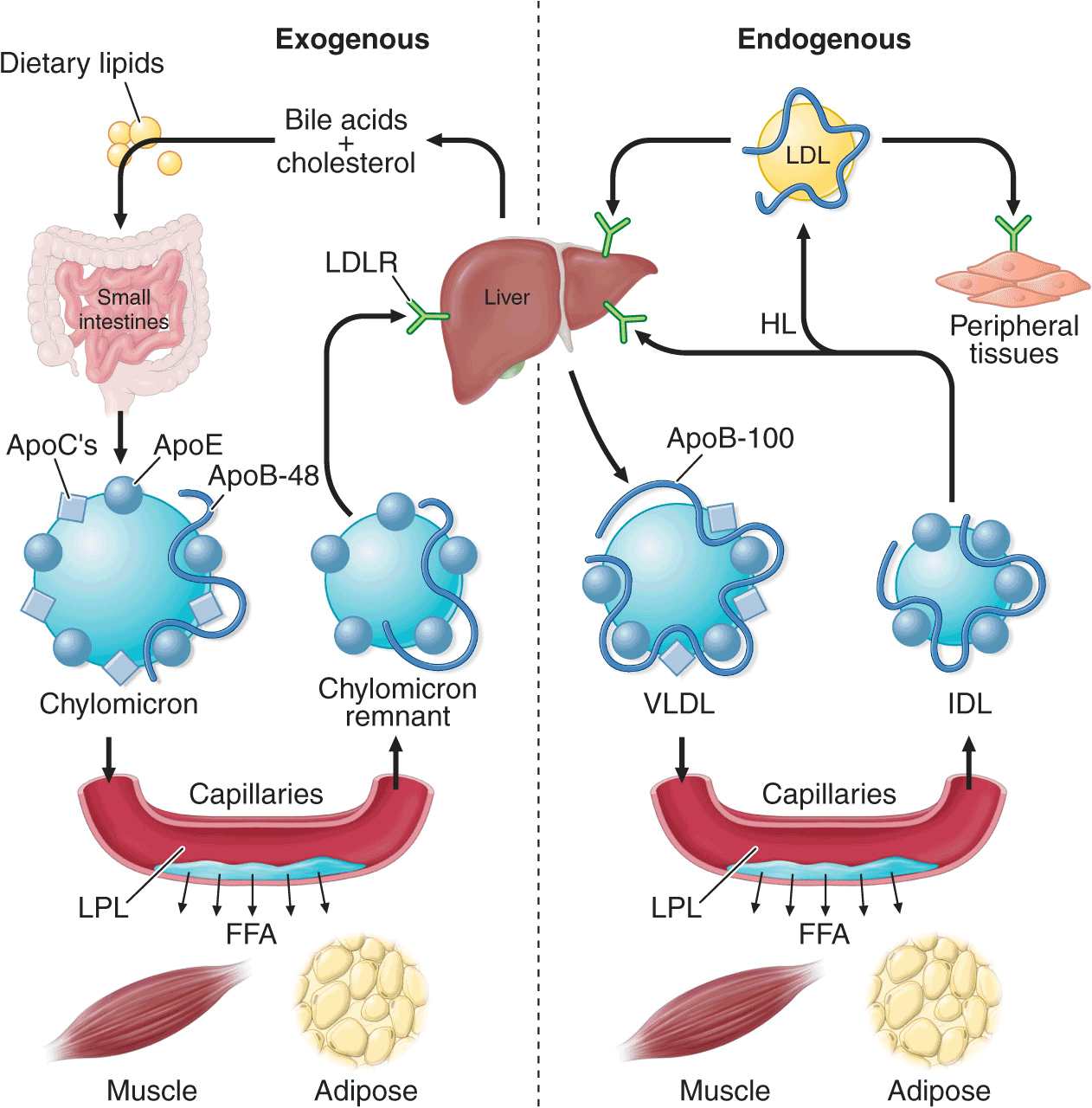
FIGURE 31-2
The exogenous and endogenous lipoprotein metabolic pathways. The exogenous pathway transports dietary lipids to the periphery and the liver. The endogenous pathway transports hepatic lipids to the periphery. LPL, lipoprotein lipase; FFA, free fatty acid; VLDL, very low-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; HL, hepatic lipase.
TRANSPORT OF HEPATIC LIPIDS (ENDOGENOUS PATHWAY)
The endogenous pathway of lipoprotein metabolism refers to the secretion of apoB-containing lipoproteins from the liver and the metabolism of these triglyceride-rich particles in peripheral tissues (Fig. 31-2). VLDL particles resemble chylomicrons in protein composition but contain apoB-100 rather than apoB-48 and have a higher ratio of cholesterol to triglyceride (~1 mg of cholesterol for every 5 mg of triglyceride). The triglycerides of VLDL are derived predominantly from the esterification of long-chain fatty acids in the liver. The packaging of hepatic triglycerides with the other major components of the nascent VLDL particle (apoB-100, cholesteryl esters, phospholipids, and vitamin E) requires the action of the enzyme microsomal triglyceride transfer protein (MTP). After secretion into the plasma, VLDL acquires multiple copies of apoE and apolipoproteins of the C series by transfer from HDL. As with chylomicrons, the triglycerides of VLDL are hydrolyzed by LPL, especially in muscle, heart, and adipose tissue. After the VLDL remnants dissociate from LPL, they are referred to as IDLs, which contain roughly similar amounts of cholesterol and triglyceride. The liver removes approximately 40–60% of IDL by LDL receptor–mediated endocytosis via binding to apoE. The remainder of IDL is remodeled by hepatic lipase (HL) to form LDL. During this process, most of the triglyceride in the particle hydrolyzed, and all apolipoproteins except apoB-100 are transferred to other lipoproteins. The cholesterol in LDL accounts for more than one-half of the plasma cholesterol in most individuals. Approximately 70% of circulating LDL is cleared by LDL receptor–mediated endocytosis in the liver. Lipoprotein(a) [Lp(a)] is a lipoprotein similar to LDL in lipid and protein composition, but it contains an additional protein called apolipoprotein(a) [apo(a)]. Apo(a) is synthesized in the liver and attached to apoB-100 by a disulfide linkage. The major site of clearance of Lp(a) is the liver, but the uptake pathway is not known.
HDL METABOLISM AND REVERSE CHOLESTEROL TRANSPORT
All nucleated cells synthesize cholesterol, but only hepatocytes and enterocytes can effectively excrete cholesterol from the body, into either the bile or the gut lumen. In the liver, cholesterol is secreted into the bile, either directly or after conversion to bile acids. Cholesterol in peripheral cells is transported from the plasma membranes of peripheral cells to the liver and intestine by a process termed “reverse cholesterol transport” that is facilitated by HDL (Fig. 31-3).
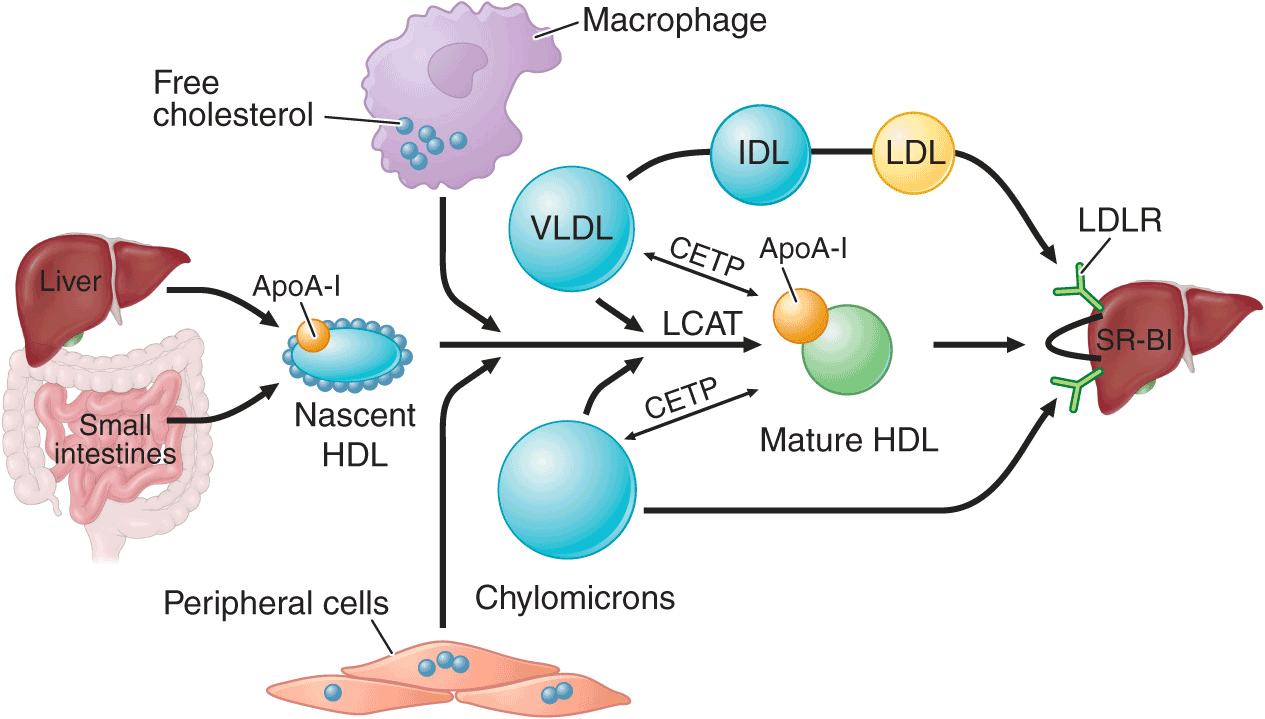
FIGURE 31-3
HDL metabolism and reverse cholesterol transport. This pathway transports excess cholesterol from the periphery back to the liver for excretion in the bile. The liver and the intestine produce nascent HDLs. Free cholesterol is acquired from macrophages and other peripheral cells and esterified by LCAT, forming mature HDLs. HDL cholesterol can be selectively taken up by the liver via SR-BI (scavenger receptor class BI). Alternatively, HDL cholesteryl ester can be transferred by CETP from HDLs to VLDLs and chylomicrons, which can then be taken up by the liver. LCAT, lecithin-cholesterol acyltransferase; CETP, cholesteryl ester transfer protein; VLDL, very low-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; HDL, high-density lipoprotein; LDLR, low-density lipoprotein receptor.
Nascent HDL particles are synthesized by the intestine and the liver. Newly secreted apoA-I rapidly acquires phospholipids and unesterified cholesterol from its site of synthesis (intestine or liver) via efflux promoted by the membrane protein ATP-binding cassette protein A1 (ABCA1). This process results in the formation of discoidal HDL particles, which then recruit additional unesterified cholesterol from the periphery. Within the HDL particle, the cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT), a plasma enzyme associated with HDL, and the more hydrophobic cholesteryl ester moves to the core of the HDL particle. As HDL acquires more cholesteryl ester it becomes spherical, and additional apolipoproteins and lipids are transferred to the particles from the surfaces of chylomicrons and VLDLs during lipolysis.
HDL cholesterol is transported to hepatocytes by both an indirect and a direct pathway. HDL cholesteryl esters can be transferred to apoB-containing lipoproteins in exchange for triglyceride by the cholesteryl ester transfer protein (CETP). The cholesteryl esters are then removed from the circulation by LDL receptor– mediated endocytosis. HDL cholesterol can also be taken up directly by hepatocytes via the scavenger receptor class B1 (SR-B1), a cell surface receptor that mediates the selective transfer of lipids to cells.
HDL particles undergo extensive remodeling within the plasma compartment by a variety of lipid transfer proteins and lipases. The phospholipid transfer protein (PLTP) has the net effect of transferring phospholipids from other lipoproteins to HDL or among different classes of HDL particles. After CETP- and PLTP-mediated lipid exchange, the triglyceride-enriched HDL becomes a much better substrate for HL, which hydrolyzes the triglycerides and phospholipids to generate smaller HDL particles. A related enzyme called endothelial lipase hydrolyzes HDL phospholipids, generating smaller HDL particles that are catabolized faster. Remodeling of HDL influences the metabolism, function, and plasma concentrations of HDL.
DISORDERS OF LIPOPROTEIN METABOLISM
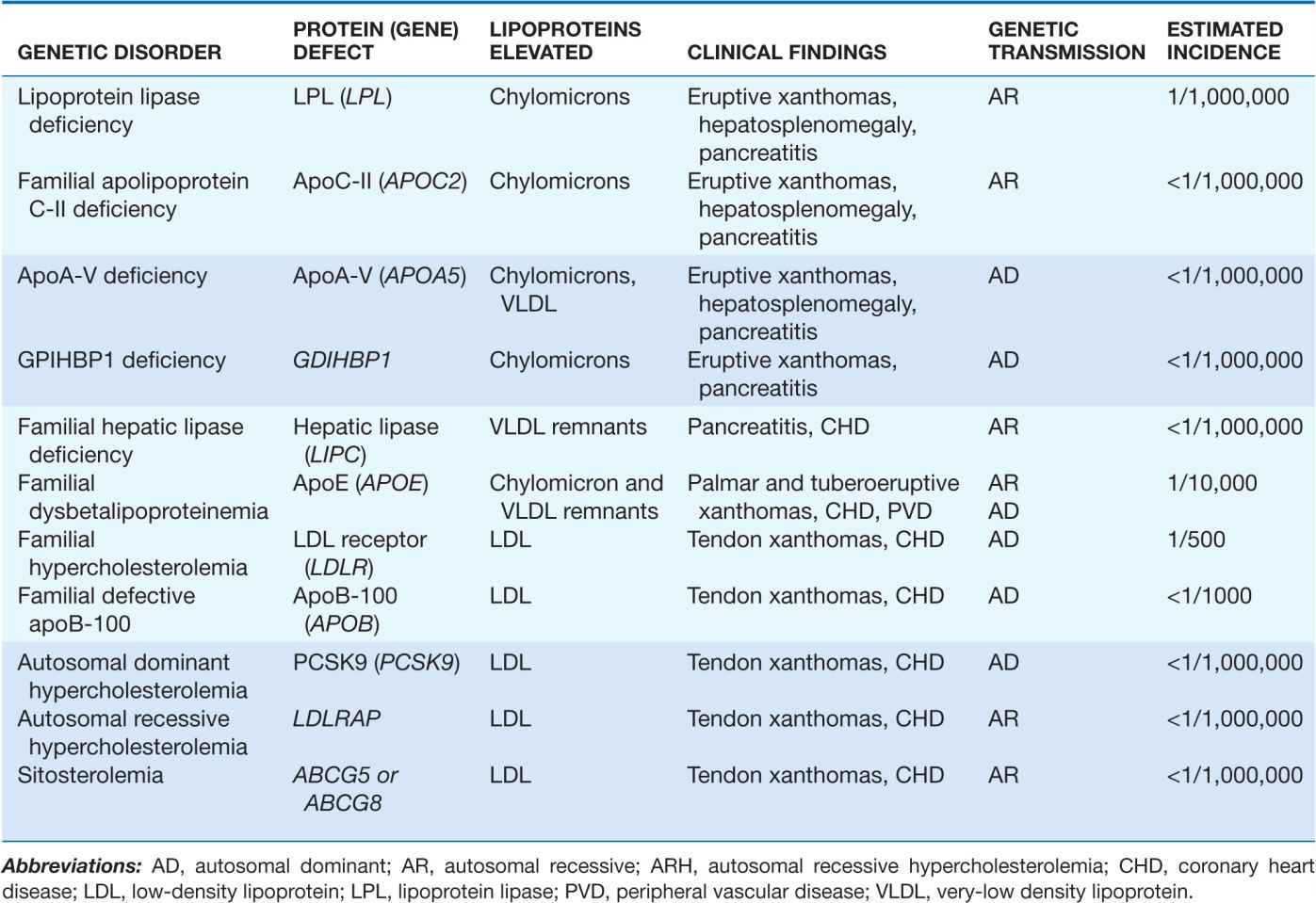
Fredrickson and Levy classified hyperlipoproteinemias according to the type of lipoprotein particles that accumulate in the blood (Type I to Type V) (Table 31-3). A classification scheme based on the molecular etiology and pathophysiology of the lipoprotein disorders complements this system and forms the basis for this chapter. The identification and characterization of genes responsible for the genetic forms of hyperlipidemia have provided important molecular insights into the critical roles of structural apolipoproteins, enzymes, and receptors in lipid metabolism (Table 31-4).
TABLE 31-3
FREDRICKSON CLASSIFICATION OF HYPERLIPOPROTEINEMIAS
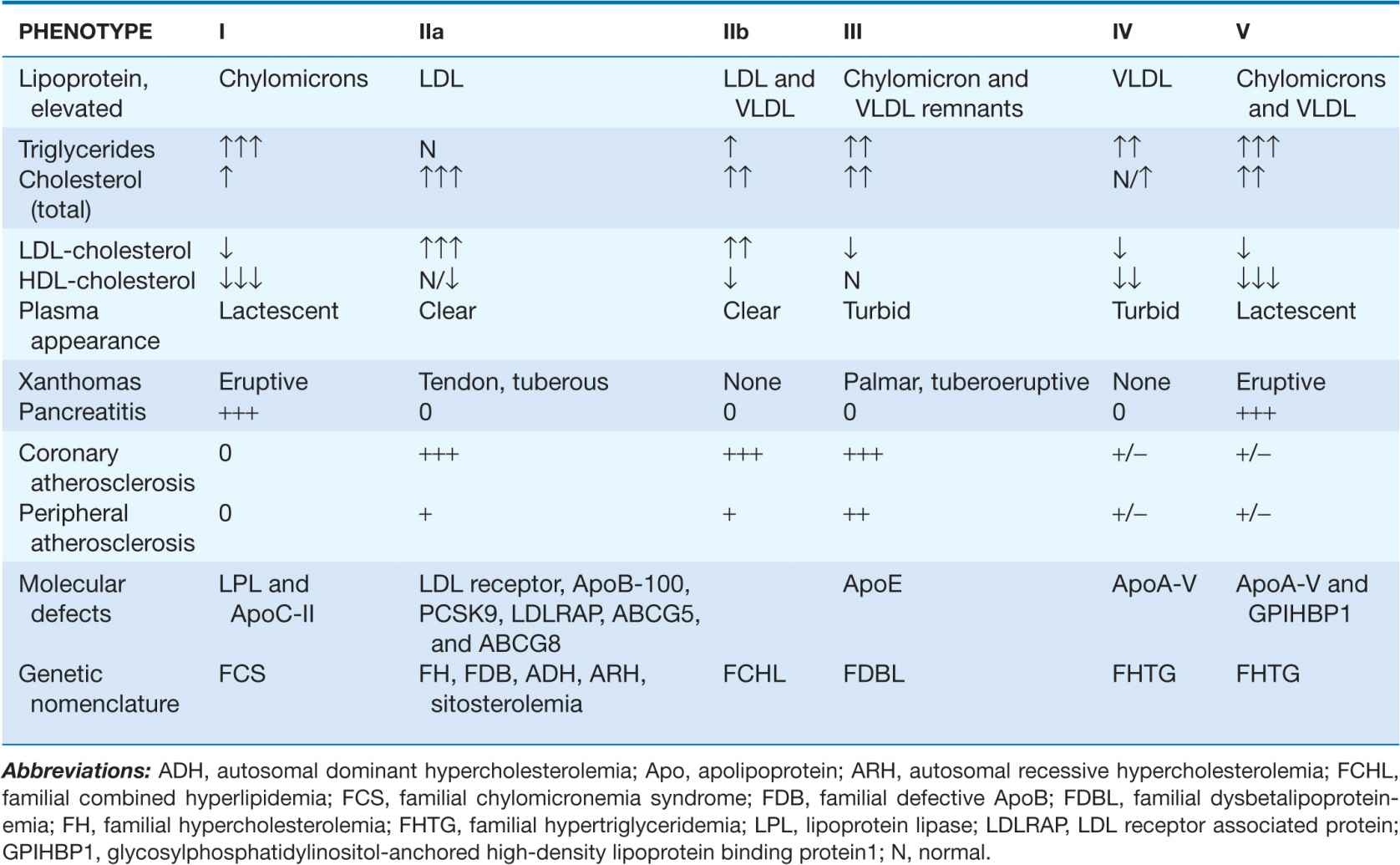
TABLE 31-4
PRIMARY HYPERLIPOPROTEINEMIAS CAUSED BY KNOWN SINGLE GENE MUTATIONS
PRIMARY DISORDERS OF ELEVATED ApoB-CONTAINING LIPOPROTEINS
A variety of genetic conditions are associated with the accumulation in plasma of specific classes of lipoprotein particles. In general, these can be divided into those causing elevated LDL-cholesterol (LDL-C) with normal triglycerides and those causing elevated triglycerides (Table 31-4).
Lipid disorders associated with elevated LDL-C and normal triglycerides
 Familial hypercholesterolemia (FH)
Familial hypercholesterolemia (FH)
FH is an autosomal codominant disorder characterized by elevated plasma levels of LDL-C with normal triglycerides, tendon xanthomas, and premature coronary atherosclerosis. FH is caused by a large number (>1000) mutations in the LDL receptor gene. It has a higher incidence in certain founder populations, such as Afrikaners, Christian Lebanese, and French Canadians. The elevated levels of LDL-C in FH are due to an increase in the production of LDL from IDL (since a portion of IDL is normally cleared by LDL receptor–mediated endocytosis) and a delayed removal of LDL from the blood. Individuals with two mutated LDL receptor alleles (FH homozygotes) have much higher LDL-C levels than those with one mutant allele (FH heterozygotes).
Homozygous FH occurs in approximately 1 in 1 million persons worldwide. Patients with homozygous FH can be classified into one of two groups based on the amount of LDL receptor activity measured in their skin fibroblasts: those patients with <2% of normal LDL receptor activity (receptor negative) and those patients with 2–25% of normal LDL receptor activity (receptor defective). Most patients with homozygous FH present in childhood with cutaneous xanthomas on the hands, wrists, elbows, knees, heels, or buttocks. Total cholesterol levels are usually >500 mg/dL and can be higher than 1000 mg/dL. The devastating complication of homozygous FH is accelerated atherosclerosis, which can result in disability and death in childhood. Atherosclerosis often develops first in the aortic root, where it can cause aortic valvular or supravalvular stenosis, and typically extends into the coronary ostia, which become stenotic. Children with homozygous FH often develop symptomatic coronary atherosclerosis before puberty; symptoms can be atypical, and sudden death is not uncommon. Untreated, receptor-negative patients with homozygous FH rarely survive beyond the second decade; patients with receptor-defective LDL receptor defects have a better prognosis but almost invariably develop clinically apparent atherosclerotic vascular disease by age 30, and often much sooner. Carotid and femoral disease develops later in life and is usually not clinically significant.
A careful family history should be taken, and plasma lipid levels should be measured in the parents and other first-degree relatives of patients with homozygous FH. The disease has >90% penetrance so both parents of FH homozygotes usually have hypercholesterolemia. The diagnosis of homozygous FH can be confirmed by obtaining a skin biopsy and measuring LDL receptor activity in cultured skin fibroblasts, or by quantifying the number of LDL receptors on the surfaces of lymphocytes using cell sorting technology. Molecular assays are also available to define the mutations in the LDL receptor by DNA sequencing. In selected populations where particular mutations predominate (e.g., Africaners and French Canadians), the common mutations can be screened for directly. Alternatively, the entire coding region needs to be sequenced for mutation detection because a large number of different LDL receptor mutations can cause disease. Ten to 15% of LDL receptor mutations are large deletions or insertions, which may be missed by routine DNA sequencing.
Combination therapy with an HMG-CoA reductase inhibitor and a second drug (cholesterol absorption inhibitor or bile acid sequestrant) sometimes reduces plasma LDL-C in those FH homozygotes who have residual LDL receptor activity, but patients with homozygous FH invariably require additional lipid-lowering therapy. Since the liver is quantitatively the most important tissue for removing circulating LDLs via the LDL receptor, liver transplantation is effective in decreasing plasma LDL-C levels in this disorder. Liver transplantation, however, is associated with substantial risks, including the requirement for long-term immunosuppression. The current treatment of choice for homozygous FH is LDL apheresis (a process by which the LDL particles are selectively removed from the circulation), which can promote regression of xanthomas and may slow the progression of atherosclerosis. Initiation of LDL apheresis should generally be delayed until approximately 5 years of age, except when evidence of atherosclerotic vascular disease is present.
Heterozygous FH is caused by the inheritance of one mutant LDL receptor allele and occurs in approximately 1 in 500 persons worldwide, making it one of the most common single-gene disorders. It is characterized by elevated plasma levels of LDL-C (usually 200–400 mg/dL) and normal levels of triglyceride. Patients with heterozygous FH have hypercholesterolemia from birth, and disease recognition is usually based on detection of hypercholesterolemia on routine screening, the appearance of tendon xanthomas, or the development of symptomatic ASCVD. Since the disease is codominant in inheritance, one parent and ~50% of the patient’s siblings usually also have hypercholesterolemia. The family history is frequently positive for premature ASCVD on one side of the family. Corneal arcus is common, and tendon xanthomas involving the dorsum of the hands, elbows, knees, and especially the Achilles tendons are present in ~75% of patients. The age of onset of ASCVD is highly variable and depends in part on the molecular defect in the LDL receptor gene and also on coexisting cardiac risk factors. FH heterozygotes with elevated plasma levels of Lp(a) appear to be at greater risk for cardiovascular complications. Untreated men with heterozygous FH have an ~50% chance of having a myocardial infarction before age 60 years. Although the age of onset of atherosclerotic heart disease is later in women with FH, coronary heart disease (CHD) is significantly more common in women with FH than in the general female population.
No definitive diagnostic test for heterozygous FH is available. Although FH heterozygotes tend to have reduced levels of LDL receptor function in skin fibroblasts, significant overlap with the LDL receptor activity levels in normal fibroblasts exists. Molecular assays are now available to identify mutations in the LDL receptor gene by DNA sequencing, but the clinical utility of pinpointing the mutation has not been demonstrated. The clinical diagnosis is usually not problematic, but it is critical that hypothyroidism, nephrotic syndrome, and obstructive liver disease be excluded before initiating therapy.
FH patients should be aggressively treated to lower plasma levels of LDL-C. Initiation of a low-cholesterol, low-fat diet is recommended, but heterozygous FH patients require lipid-lowering drug therapy. Statins are effective in heterozygous FH, but combination drug therapy with the addition of a cholesterol absorption inhibitor and/or bile acid sequestrant is frequently required, and the addition of nicotinic acid is sometimes needed. Heterozygous FH patients who cannot be adequately controlled on combination drug therapy are candidates for LDL apheresis.
Familial defective ApoB-100 (FDB)
FDB is a dominantly inherited disorder that clinically resembles heterozygous FH. The disease is rare in most populations except individuals of German descent, where the frequency can be as high as 1 in 1000. FDB is characterized by elevated plasma LDL-C levels with normal triglycerides, tendon xanthomas, and an increased incidence of premature ASCVD. FDB is caused by mutations in the LDL receptor–binding domain of apoB-100, most commonly due to a substitution of glutamine for arginine at position 3500. As a consequence of the mutation in apoB-100, LDL binds the LDL receptor with reduced affinity, and LDL is removed from the circulation at a reduced rate. Patients with FDB cannot be clinically distinguished from patients with heterozygous FH, although patients with FDB tend to have lower plasma levels of LDL-C than FH heterozygotes. The apoB-100 gene mutation can be detected directly, but genetic diagnosis is not currently encouraged since the recommended management of FDB and heterozygous FH is identical.
 Autosomal dominant hypercholesterolemia due to mutations in PCSK9 (ADH-PCSK9 or ADH3)
Autosomal dominant hypercholesterolemia due to mutations in PCSK9 (ADH-PCSK9 or ADH3)
ADH-PCSK9 is a rare autosomal dominant disorder caused by gain-of-function mutations in proprotein convertase subtilisin/kexin type 9 (PCSK9). PCSK9 is a secreted protein that binds to the LDL receptor, resulting in its degradation. Normally, after LDL binds to the receptor it is internalized along with the receptor. In the low pH of the endosome, LDL dissociates from the receptor and returns to the cell surface. The LDL is delivered to the lysosome. When PCSK9 binds the receptor, the complex is internalized and the receptor is redirected to the lysosome rather than to the cell surface. The missense mutations in PCSK9 that cause hypercholesterolemia enhance the activity of PCSK9. As a consequence, the number of hepatic LDL receptors is reduced. Patients with ADH-PCSK9 are indistinguishable clinically from patients with FH. Interestingly, loss-of-function mutations in PCSK9 cause low LDL-C levels (see later).
 Autosomal recessive hypercholesterolemia (ARH)
Autosomal recessive hypercholesterolemia (ARH)
ARH is a rare disorder (except in Sardinia, Italy) due to mutations in a protein (ARH, also called LDLR adaptor protein, LDLRAP) involved in LDL receptor–mediated endocytosis in the liver. In the absence of LDLRAP, LDL binds to the LDL receptor but the lipoprotein-receptor complex fails to be internalized. ARH, like homozygous FH, is characterized by hypercholesterolemia, tendon xanthomas, and premature coronary artery disease (CAD). The levels of plasma LDL-C tend to be intermediate between the levels present in FH homozygotes and FH heterozygotes, and CAD is not usually symptomatic until at least the third decade. LDL receptor function in cultured fibroblasts is normal or only modestly reduced in ARH, whereas LDL receptor function in lymphocytes and the liver is negligible. Unlike FH homozygotes, the hyperlipidemia responds partially to treatment with HMG-CoA reductase inhibitors, but these patients usually require LDL apheresis to lower plasma LDL-C to recommended levels.
 Sitosterolemia
Sitosterolemia
Sitosterolemia is another rare autosomal recessive disease that can result in severe hypercholesterolemia, tendon xanthomas, and premature ASCVD. Sitosterolemia is caused by mutations in either of two members of the ATP-binding cassette (ABC) half transporter family, ABCG5 and ABCG8. These genes are expressed in enterocytes and hepatocytes. The proteins heterodimerize to form a functional complex that pumps plant sterols such as sitosterol and campesterol, and animal sterols, predominantly cholesterol, into the gut lumen and into the bile. In normal individuals, <5% of dietary plant sterols are absorbed by the proximal small intestine and delivered to the liver. Absorbed plant sterols are preferentially secreted into the bile and are maintained at very low levels. In sitosterolemia, the intestinal absorption of sterols is increased and biliary excretion of the sterols is reduced, resulting in increased plasma and tissue levels of both plant sterols and cholesterol.
Incorporation of plant sterols into cell membranes results in misshapen red blood cells and megathrombocytes that are visible on blood smear. Episodes of hemolysis are a distinctive clinical feature of this disease compared to other genetic forms of hypercholesterolemia.
Sitosterolemia is diagnosed by demonstrating an increase in the plasma level of sitosterol using gas chromatography. The hypercholesterolemia is unusually responsive to reductions in dietary cholesterol content and should be suspected in individuals who have a >40% reduction in plasma cholesterol level on a lowcholesterol diet. The hypercholesterolemia does not respond to HMG-CoA reductase inhibitors, whereas bile acid sequestrants and cholesterol-absorption inhibitors such as ezetimibe, are effective in reducing plasma sterol levels in these patients.
 Polygenic hypercholesterolemia
Polygenic hypercholesterolemia
This condition is characterized by hypercholesterolemia due to elevated LDL-C with a normal plasma level of triglyceride in the absence of secondary causes of hypercholesterolemia. Plasma LDL-C levels are generally not as elevated as they are in FH and FDB. Family studies are useful to differentiate polygenic hypercholesterolemia from the single-gene disorders described above; one-half of the first-degree relatives of patients with FH and FDB are hypercholesterolemic, whereas <10% of first-degree relatives of patients with polygenic hypercholesterolemia have hypercholesterolemia. Treatment of polygenic hypercholesterolemia is identical to that of other forms of hypercholesterolemia.
Elevated plasma levels of lipoprotein(a)
Unlike the other major classes of lipoproteins, that have a normal distribution in the population, plasma levels of Lp(a) have a highly skewed distribution with levels varying over a 1000-fold range. Levels are strongly influenced by genetic factors, with individuals of African and South Asian descent having higher levels than those of European descent. Although it has been well documented that elevated levels of Lp(a) are associated with an increase in ASCVD, lowering plasma levels of Lp(a) has not been demonstrated to reduce cardiovascular risk.
Lipid disorders associated with elevated triglycerides
 Familial chylomicronemia syndrome (Type I hyperlipoproteinemia; lipoprotein lipase, and ApoC-II deficiency)
Familial chylomicronemia syndrome (Type I hyperlipoproteinemia; lipoprotein lipase, and ApoC-II deficiency)
As noted above, LPL is required for the hydrolysis of triglycerides in chylomicrons and VLDLs, and apoC-II is a cofactor for LPL (Fig. 31-2). Genetic deficiency or inactivity of either protein results in impaired lipolysis and profound elevations in plasma chylomicrons. These patients can also have elevated plasma levels of VLDL, but chylomicronemia predominates. The fasting plasma is turbid, and if left at 4°C (39.2°F) for a few hours, the chylomicrons float to the top and form a creamy supernatant. In these disorders, called familial chylomicronemia syndromes, fasting triglyceride levels are almost invariably >1000 mg/dL. Fasting cholesterol levels are also elevated but to a lesser degree.
LPL deficiency has autosomal recessive inheritance and has a frequency of approximately 1 in 1 million in the population. ApoC-II deficiency is also recessive in inheritance pattern and is even less common than LPL deficiency. Multiple different mutations in the LPL and apoC-II genes cause these diseases. Obligate LPL heterozygotes have normal or mild-to-moderate elevations in plasma triglyceride levels, whereas individuals heterozygous for mutation in apoC-II do not have hypertriglyceridemia.
Both LPL and apoC-II deficiency usually present in childhood with recurrent episodes of severe abdominal pain due to acute pancreatitis. On funduscopic examination, the retinal blood vessels are opalescent (lipemia retinalis). Eruptive xanthomas, which are small, yellowish-white papules, often appear in clusters on the back, buttocks, and extensor surfaces of the arms and legs. These typically painless skin lesions may become pruritic. Hepatosplenomegaly results from the uptake of circulating chylomicrons by reticuloendothelial cells in the liver and spleen. For unknown reasons, some patients with persistent and pronounced chylomicronemia never develop pancreatitis, eruptive xanthomas, or hepatosplenomegaly. Premature CHD is not generally a feature of familial chylomicronemia syndromes.
The diagnoses of LPL and apoC-II deficiency are established enzymatically in specialized laboratories by assaying triglyceride lipolytic activity in postheparin plasma. Blood is sampled after an IV heparin injection to release the endothelial-bound LPL. LPL activity is profoundly reduced in both LPL and apoC-II deficiency; in patients with apoC-II deficiency, it normalizes after the addition of normal plasma (providing a source of apoC-II). Molecular sequencing of the genes can be used to confirm the diagnosis.
The major therapeutic intervention in familial chylomicronemia syndromes is dietary fat restriction (to as little as 15 g/d) with fat-soluble vitamin supplementation. Consultation with a registered dietician familiar with this disorder is essential. Caloric supplementation with medium-chain triglycerides, which are absorbed directly into the portal circulation, can be useful but may be associated with hepatic fibrosis if used for prolonged periods. If dietary fat restriction alone is not successful in resolving the chylomicronemia, fish oils have been effective in some patients. In patients with apoC-II deficiency, apoC-II can be provided by infusing fresh-frozen plasma to resolve the chylomicronemia in the acute setting. Management of patients with familial chylomicronemia syndrome is particularly challenging during pregnancy when VLDL production is increased and may require plasmapheresis to remove the circulating chylomicrons.
 ApoA-V deficiency
ApoA-V deficiency
Another apolipoprotein, apoA-V, circulates at much lower concentrations than the other major apolipoproteins. Individuals harboring mutations in both apoA-V alleles can present as adults with chylomicronemia. The exact mechanism of action of apoA-V is not known, but it appears to be required for the association of VLDL and chylomicrons with LPL.
 GPIHBP1 deficiency
GPIHBP1 deficiency
After LPL is synthesized in adipocytes, myocytes, or other cells, it is transported across the vascular endothelium and is attached to a protein on the endothelial surface of capillaries called GPIHBP1. Homozygosity for mutations that interfere with GPIHBP1 synthesis or folding cause severe hypertriglyceridemia. The frequency of chylomicronemia due to mutations in GHIHBP1 has not been established but appears to be very rare.
 Hepatic lipase deficiency
Hepatic lipase deficiency
HL is a member of the same gene family as LPL and hydrolyzes triglycerides and phospholipids in remnant lipoproteins and HDLs. HL deficiency is a very rare autosomal recessive disorder characterized by elevated plasma levels of cholesterol and triglycerides (mixed hyperlipidemia) due to the accumulation of circulating lipoprotein remnants and either a normal or elevated plasma level of HDL-C. The diagnosis is confirmed by measuring HL activity in postheparin plasma. Due to the small number of patients with HL deficiency, the association of this genetic defect with ASCVD is not clearly known, but lipid-lowering therapy is recommended.
 Familial dysbetalipoproteinemia (Type III hyperlipoproteinemia)
Familial dysbetalipoproteinemia (Type III hyperlipoproteinemia)
Like HL deficiency, familial dysbetalipoproteinemia (FDBL) (also known as Type III hyperlipoproteinemia or familial broad β disease) is characterized by a mixed hyperlipidemia due to the accumulation of remnant lipoprotein particles. ApoE is present in multiple copies on chylomicron and VLDL remnants and mediates their removal via hepatic lipoprotein receptors (Fig. 31-2). FDBL is due to genetic variations in apoE that interfere with its ability to bind lipoprotein receptors. The APOE gene is polymorphic in sequence, resulting in the expression of three common isoforms: apoE3, which is the most common; and apoE2 and apoE4, which both differ from apoE3 by a single amino acid. Although associated with slightly higher LDL-C levels and increased CHD risk, the apoE4 allele is not associated with FDBL. Patients with apoE4 have an increased incidence of late-onset Alzheimer’s disease. ApoE2 has a lower affinity for the LDL receptor; therefore, chylomicron and VLDL remnants containing apoE2 are removed from plasma at a slower rate. Individuals who are homozygous for the E2 allele (the E2/E2 genotype) comprise the most common subset of patients with FDBL.
Approximately 0.5% of the general population are apoE2/E2 homozygotes, but only a small minority of these individuals develop FDBL. In most cases, an additional, identifiable factor precipitates the development of hyperlipoproteinemia. The most common precipitating factors are a high-fat diet, diabetes mellitus, obesity, hypothyroidism, renal disease, HIV infection, estrogen deficiency, alcohol use, or certain drugs. Other mutations in apoE can cause a dominant form of FDBL where the hyperlipidemia is fully manifest in the heterozygous state, but these mutations are rare.
Patients with FDBL usually present in adulthood with incidental hyperlipidemia, xanthomas, premature coronary disease, or peripheral vascular disease. The disease seldom presents in women before menopause. Two distinctive types of xanthomas, tuberoeruptive and palmar, are seen in FDBL patients. Tuberoeruptive xanthomas begin as clusters of small papules on the elbows, knees, or buttocks and can grow to the size of small grapes. Palmar xanthomas (alternatively called xanthomata striata palmaris) are orange-yellow discolorations of the creases in the palms and wrists. In FDBL, in contrast to other disorders of elevated triglycerides, the plasma levels of cholesterol and triglyceride are often elevated to a similar degree and the level of HDL-C is usually normal rather than being low.
The traditional approaches to diagnosis of this disorder are lipoprotein electrophoresis (broad β band) or ultracentrifugation (ratio of VLDL-C to total plasma triglyceride >0.30). Protein methods (apoE phenotyping) or DNA-based methods (apoE genotyping) can be performed to confirm homozygosity for apoE2. However, absence of the apoE2/E2 genotype does not rule out the diagnosis of FDBL, since other mutations in apoE can cause this condition.
Since FDBL is associated with increased risk of premature ASCVD, it should be treated aggressively. Subjects with FDBL tend to have more peripheral vascular disease than is typically seen in FH. Other metabolic conditions that can worsen the hyperlipidemia (see earlier) should be aggressively treated. Patients with FDBL are typically very diet responsive and can respond favorably to weight reduction and to low-cholesterol, low-fat diets. Alcohol intake should be curtailed. HMGCoA reductase inhibitors, fibrates, and niacin are all generally effective in the treatment of FDBL, and sometimes combination drug therapy is required.
 Familial hypertriglyceridemia (FHTG)
Familial hypertriglyceridemia (FHTG)
FHTG is a relatively common (~1 in 500) autosomal dominant disorder of unknown etiology characterized by moderately elevated plasma triglycerides accompanied by more modest elevations in cholesterol. Since the major class of lipoproteins elevated in this disorder is VLDL, patients with this disorder are often referred to as having Type IV hyperlipoproteinemia (Fredrickson classification, Table 31-3). The elevated plasma levels of VLDL are due to increased production of VLDL, impaired catabolism of VLDL, or a combination of these mechanisms. Some patients with FHTG have a more severe form of hyperlipidemia in which both VLDLs and chylomicrons are elevated (Type V hyperlipidemia), since these two classes of lipoproteins compete for the same lipolytic pathway. Increased intake of simple carbohydrates, obesity, insulin resistance, alcohol use, and estrogen treatment, all of which increase VLDL synthesis, can exacerbate this syndrome. FHTG appears not to be associated with increased risk of ASCVD in many families.
The diagnosis of FHTG is suggested by the triad of elevated levels of plasma triglycerides (250–1000 mg/dL), normal or only mildly increased cholesterol levels (<250 mg/dL), and reduced plasma levels of HDL-C. Plasma LDL-C levels are generally not increased and are often reduced due to defective metabolism of the triglyceride-rich particles. The identification of other first-degree relatives with hypertriglyceridemia is useful in making the diagnosis. FDBL and familial combined hyperlipidemia (FCHL) should also be ruled out since these two conditions are associated with a significantly increased risk of ASCVD. The plasma apoB levels are lower and the ratio of plasma triglyceride to cholesterol is higher in FHTG than in either FDBL or FCHL.
It is important to consider and rule out secondary causes of hypertriglyceridemia (Table 31-5) before making the diagnosis of FHTG. Lipid-lowering drug therapy can frequently be avoided with appropriate dietary and lifestyle changes. Patients with plasma triglyceride levels >500 mg/dL after a trial of diet and exercise should be considered for drug therapy to avoid the development of chylomicronemia and pancreatitis. Fibrate drugs or fish oils (omega 3 fatty acids) are reasonable first-line approaches for FHTG, and niacin can also be considered in this condition. For more moderate elevations in triglyceride levels (250–500 mg/dL), statins are effective at lowering triglyceride levels.
TABLE 31-5
SECONDARY FORMS OF HYPERLIPIDEMIA
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


