and Alessandra Cancellieri2
(1)
Department of Radiology, Bellaria Hospital, Bologna, Italy
(2)
Department of Pathology, Maggiore Hospital, Bologna, Italy
Radiology | Maurizio Zompatori Domenico Attinà |
BHD | Birt–Hogg–Dubè | Page 212 |
Bronchiectasis, cystic | Bronchiectasis, cystic | Page 214 |
CF | Cystic fibrosis | Page 216 |
CPFE | Combined pulmonary fibrosis and emphysema | Page 218 |
Emphysema, CL & PS | Emphysema, centrilobular and paraseptal | Page 220 |
LAM | Lymphangioleiomyomatosis | Page 222 |
LCH, advanced | Langerhans cell histiocytosis, advanced | Page 224 |
LIP | Lymphocytic interstitial pneumonia | Page 226 |
LTB papillomatosis | Laryngotracheobronchial papillomatosis | Page 228 |
Metastases, cystic | Metastases, cystic | Page 230 |
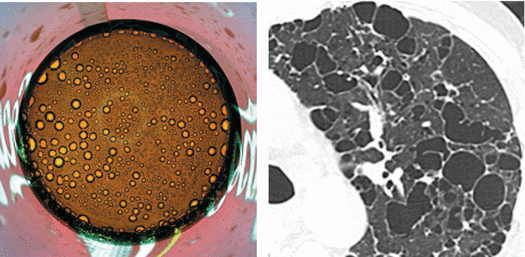
Birt–Hogg–Dubè (BHD)
Definition
The syndrome was reported for the first time in 1977 by Birt, Hogg, and Dubè who described small papular skin lesions distributed on the scalp, forehead, face, and neck in 15 out of 70 members of the same family. Histologic examination of the lesions reveals fibrofolliculomas, trichodiscomas, and acrochordons. Birt–Hogg–Dubè syndrome is a rare autosomal dominant genodermatosis caused by a mutation in the FLCN gene, which encodes for tumor suppressor protein, folliculin. Mutation predisposes to the development of cutaneous hamartomas (particularly fibrofolliculomas), cystic lung lesions, pneumothorax, and renal tumors ranging from benign oncocytoma to renal cell cancer. Skin lesions, however, may be absent.
BHD, Hornstein–Knickenberg syndrome, fibrofolliculomas with trichodiscomas and acrochordons
Birt AR, Hogg GR, Dubé WJ (1977) Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 113(12):1674
Souza CA (2005) Birt-Hogg-Dubé syndrome: a rare cause of pulmonary cysts. AJR Am J Roentgenol 185(5):1237
High-Resolution CT: HRCT
Key Signs
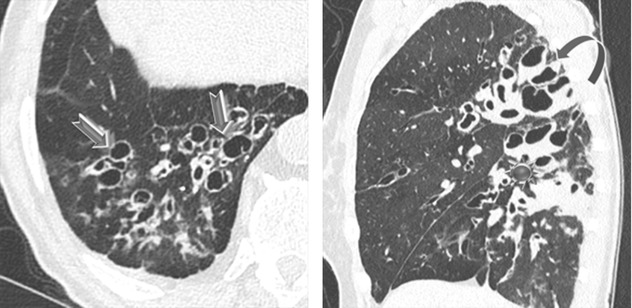
Thin-walled, sometimes septated cysts, few in number, and variable in size (from a few millimeters to several centimeters) (➨); the cysts may be rounded or lenticular in shape.
Distribution
Lower and medial zone predominance, often subpleural in location (90 %) (►)
Cysts are often larger than those in LAM and LCH. Subpleural cysts, which may involve the fissures, are more frequent than in other cystic diseases.
Agarwal PP (2011) Thoracic CT findings in Birt-Hogg-Dubè syndrome. AJR Am J Roentgenol 196:349
Ancillary Signs
Absent
Non-parenchymal Signs
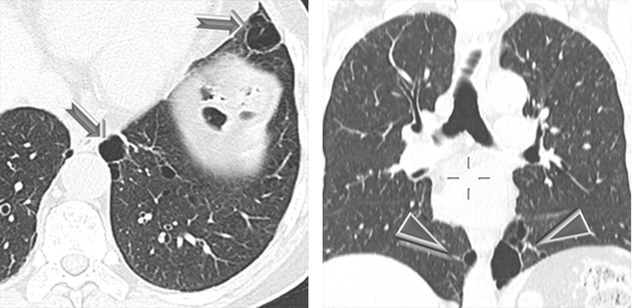
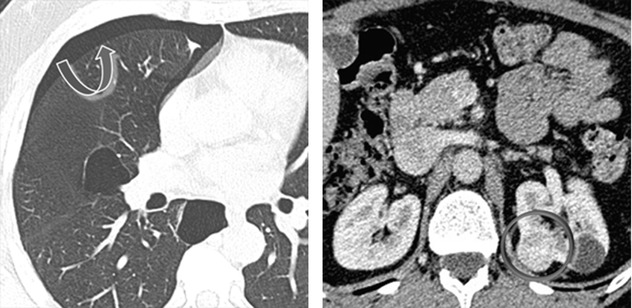
Pneumothorax due to rupture of subpleural cysts ( )
)
Renal tumors, often multiple and bilateral ( )
)
Patients aged 20–40 years frequently develop pneumothorax, usually without prior diagnosis of the underlying genetic syndrome, and may have isolated lung cysts without involvement of skin and/or kidney.
Renal tumors include hybrid chromophobe oncocytoma (50 %), chromophobe carcinomas (34 %), clear cell carcinomas (9 %), oncocytomas (5 %), and papillary renal cell cancers (2 %). Extrarenal neoplasm has been occasionally observed.
Menko FH (2009) Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol 10(12):1199
Tomassetti S (2011) Pulmonary features of Birt-Hogg-Dubé syndrome: cystic lesions and pulmonary histiocytoma. Respir Med 105:768
Course and Complications
Often recurrent pneumothorax
Dal Sasso AA (2015) Birt-Hogg-Dubé syndrome. State-of-the-art review with emphasis on pulmonary involvement. Respir Med 109(3):289
Bronchiectasis, Cystic
Definition
Bronchiectasis refers to an abnormal, permanent, and irreversible dilatation of the bronchial tree due to the destruction of the elastic and muscular components of bronchial walls. This condition represents the final common pathway of several respiratory and systemic diseases. Cystic bronchiectasis is the most severe manifestation of bronchial dilation. Smaller dilations with a cylindrical or varicose appearance may coexist. Bronchiectasis typically presents with chronic productive cough, recurrent chest infections, and hemoptysis (50 % of cases). The disease is more commonly encountered in middle aged and elderly.
Clustered cysts
Barker AF (2002) Bronchiectasis. N Engl J Med 346:1383
High-Resolution CT: HRCT
Key Signs
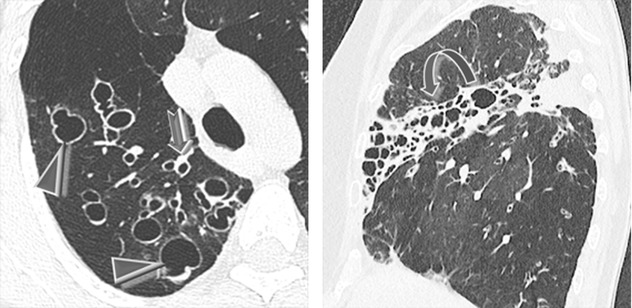
Cystic bronchiectasis is arranged in grapelike clusters (►), often around a stem being the bronchovascular pedicle (bunch of grapes sign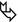 ).
).
Bronchial wall thickening.
Distribution
Focal or diffuse, depending on the underlying disease. Upper predominance is common in cystic fibrosis (CF) and allergic bronchopulmonary aspergillosis (ABPA). Cartilage deficiency disorders (Mounier–Kuhn and Williams–Campbell syndrome) are characterized by central bronchiectases.
Cystic or varicose bronchiectasis can be confused with parenchymal cystic disease, because dilated airways may show a cystic-like appearance on cross-sectional views. Assessment of adjacent sections on CT and multiplanar reconstructions should differentiate between these conditions.
Hansell DM (1998) Bronchiectasis. Radiol Clin North Am 36:107
Cantin L (2009) Bronchiectasis AJR Am J Roentgenol 193:W158
Milliron B (2015) Bronchiectasis: mechanisms and imaging clues of associated common and uncommon diseases. Radiographics 35(4):1011
Ancillary Signs
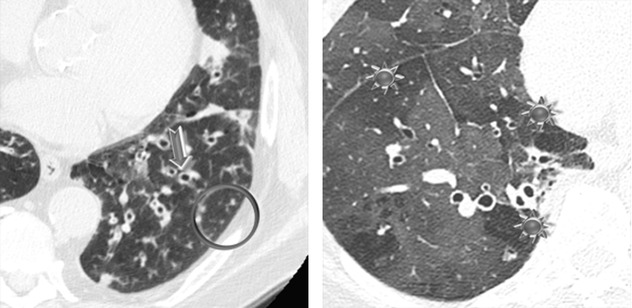
Varicoid and cylindrical bronchiectasis (signet ring sign ➨)
Mucus plugging within bronchi (finger in glove sign) (please also refer this sign in the “Case-Based Glossary with Tips and Tricks”)
Centrilobular nodules and tree-in-bud pattern related to cellular bronchiolitis
Air trapping due to obliterative bronchiolitis ( )
)
Air-fluid levels (mucus, pus, or blood)
Inflammatory consolidations
Non-parenchymal Signs
Enlarged mediastinal lymph nodes
Dilated and dysmorphic appearance of the trachea (tracheomegaly) and the main bronchi in cases of Mounier–Kuhn syndrome
Pathophysiology consists in a vicious cycle which includes chronic inflammation and inability to clear mucoid secretions. A predisposed individual develops a robust inflammatory response to pulmonary infection or tissue injury. The inflammation that results is partially responsible for the structural damage to the airways. The structural abnormalities allow for mucus stasis, which favors continued chronic infection, and the vicious cycle persists.
McShane PJ (2013) Non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med 188:647
Course and Complications
Recurrent infection, especially caused by nontuberculous mycobacterial species, Pseudomonas aeruginosa and Haemophilus influenzae, with frequent hospitalizations and increased risk of death, can occur.
Cystic lesions may extend progressively especially in relation to the recurrence of inflammatory events and phenomena of scarring with retraction.
Inclusions inside the cysts (mycetoma).
An intracystic macronodule/mass is often due to a mycetoma (particularly by Aspergillus colonization); please also refer to air crescent sign in the Case-based glossary. Rarely, the cause of intracystic macronodule/mass is neoplastic; however, only a mycetoma moves when the patient’s position is changed. Sometimes, the cysts may be completely filled with material and assume a pseudo-nodular appearance.
McGuinness G (2002) CT of airways disease and bronchiectasis. Radiol Clin North Am 40:1
Cystic Fibrosis (CF)
Definition
Cystic fibrosis (CF) is an autosomal recessive genetic disease which affects the exocrine function of the lungs, liver, pancreas, and small bowel resulting in progressive disability and multisystem failure. CF is more common in children and young adults of Caucasian origin. Mutation in the CF transmembrane regulator (CFTR) gene alters chloride passage across cell membranes. In the lungs it causes an abnormally low water content in the airway secretions, resulting in reduced clearance, mucus plugs, and increased incidence of airways infections. Although a few tens of mutations are responsible for most cases of CF, nearly 2000 CFTR gene mutations have been identified. These genotypic variants often cause a less severe disease and therefore in these patients CF is more likely to be detected in adulthood.
CF, Mucoviscidosis
O’Sullivan BP (2009) Cystic fibrosis. Lancet 373:1891
High-Resolution CT: HRCT
Key Signs
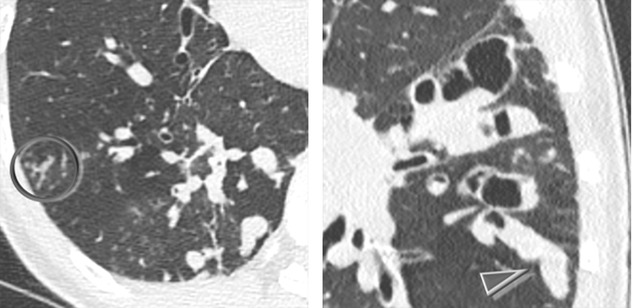
Extensive cystic bronchiectases appearing as “clustered cysts” often found adjacent to the bronchial tree (bunch of grapes sign ➨)
Cystic spaces within dense fibrotic and postinfectious conglomerates ( )
)
Air-fluid levels in the dilated cystic spaces (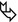 )
)
Bronchial wall and peribronchial interstitial thickening
Distribution
Upper lobe predominance of findings is seen in many but not all cases; a diffuse distribution is also a common finding.
Bronchiectasis begins as cylindrical and progresses through varicoid to cystic forms. It can be filled with air, mucus, or exudate (due to infection). Structural changes detected by HRCT scans often precede functional changes in children with CF.
The lungs of CF patients are frequently colonized with a variety of characteristic organisms such as Pseudomonas aeruginosa, Staphylococcus aureus, Aspergillus fumigatus, and nontuberculous mycobacterial species. Aspergillus may cause an asthmatic condition called allergic bronchopulmonary aspergillosis (ABPA), characterized by an exaggerated response of the immune system to the fungus spores, which lead, if not treated, to a more rapid functional impairment.
Dodd JD (2015) Imaging in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin Respir Crit Care Med 36:194
Mott LS (2012) Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 67:509
Ancillary Signs
Extensive cylindrical bronchiectasis
A mosaic pattern of attenuation secondary to air trapping due to obstructed bronchi and bronchioles.
Commonly seen tree-in-bud pattern ( ) indicates the diffuse bronchiolitis that typically occurs in cystic fibrosis.
) indicates the diffuse bronchiolitis that typically occurs in cystic fibrosis.
Consolidations (infections).
Mucus plugging within bronchi (finger in glove sign ►) (please refer to finger in glove sign in the “Case-Based Glossary with Tips and Tricks”).
Non-parenchymal Signs
Lymph node enlargement
Chronic cor pulmonale in the advanced stages of disease
Hara AK (2009) Iterative reconstruction technique for reducing body radiation dose at CT: feasibility study. AJR Am J Roentgenol 193:764
Course and Complications
Recurrent infectious bronchiolitis and lung function impairment.
The material in the cystic spaces has a fluid or superfluid density (pus) which remains unchanged in the presence of contrast material. Isolated opacities of variable density (mycetomas, clots, dense mucus), which move with the patient’s position, may also be seen.
Pulmonary arterial hypertension and chronic cor pulmonale.
As a result of the improvement of medical care and early detection, the average life expectancy of patients with CF has been steadily increasing and quality of life has improved. Several CF HRCT scoring systems have been developed to monitor the extension of lung damage, compare clinical severity, evaluate the effects of therapeutic interventions, and estimate prognosis.
Recent advances in the therapy of CF have improved the survival rate. Low-dose CT techniques should be considered to minimize radiation exposure, as these patients may have numerous CT for the long-term follow-up of lung disease. These techniques demonstrated the same accuracy to identify CF exacerbations as the high-dose techniques.
Ng MY (2014) Pulmonary complications of cystic fibrosis. Clin Radiol 69:e153
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


