Diffuse Lung Disease
Francis C. Nichols
Sebastian Defranchi
Diffuse parenchymal lung disease (DPLD) comprises a wide range of heterogenous lung disorders characterized by parenchymal lung injury with varying degrees of inflammation and fibrosis. Since Hamman and Rich35 described rapidly progressive pulmonary fibrosis, more than 100 disorders have been classified as diffuse diseases. DPLDs encompass disorders of known cause as well as others of unknown cause.105 Identifiable causes include infectious, inflammatory, malignant, drug-related, occupational/environmental, and other conditions; the etiology of most DPLDs is idiopathic. Among the DPLDs of unknown cause are idiopathic interstitial pneumonias (IIP), granulomatous lung disorders (e.g., sarcoidosis), and other forms of interstitial lung disease (ILD) including lymphangioleiomyomatosis (LAM), pulmonary Langerhans cell histiocytosis X (HX), and eosinophilic pneumonia. Some of the causes of DPLD and the diseases associated with them are shown in Figure 97-1, and common abbreviations are shown in Table 97-1. With the exceptions of lung transplantation, which is necessary in only the minority of DPLD patients, the occasional request for tumor resection, or the treatment of pulmonary complications such as pneumothorax and pleural effusion, the role of the thoracic surgeon in DPLD is primarily diagnostic. While advances in clinicopathologic correlation have lessened the need for surgical biopsy, in many instances the need for definitive tissue sampling persists, given the limitations of small specimens that hamper pathologic diagnosis. When biopsy is considered, the surgeon should serve not solely as a technician but also as an adviser in determining the timing, method, and wisdom of invasive diagnostic efforts.
Pathogenesis of Fibrosis in the Lung
The microscopic anatomy and cell populations of the lung are discussed in Chapters 3 and 4. DPLD involves the gas-exchanging structures, including bronchioles, alveolar ducts, alveolar sacs, interstitium, and small vessels. The precise molecular mechanisms underlying the development of fibrosis and leading to irreversible lung parenchymal destruction are still unknown.96 It is believed that the fibrotic pulmonary process begins with damage to the alveolar epithelial cells. These damaged cells, in turn, secrete many profibrotic substances that lead to fibroblast recruitment and ultimately to an abnormal reparative process that finishes with irreversible fibrosis.
Many cell types are involved in the pathogenesis of pulmonary fibrosis. These include type I and type II pneumonocytes (alveolar epithelial cells), endothelial cells, fibroblasts, alveolar macrophages, and lymphocytes as well as many other cells. In the adult, type I and II pneumonocytes constitute the epithelia of the lung alveoli. Type I pneumonocytes cover >90% of the alveolar surface, they have a minimal thickness and are the cells specialized in gas exchange. Type II cells are rounded, secrete surfactant, are involved in sodium transportation, serve as progenitor cells for type I cells, and are involved in the immune response. Type I cells are more vulnerable to injury while type II are highly resistant. Most of the current models that explain the pathologic fibrotic process in the lung center on the theory of injury to the epithelial component of the alveoli. Injury to the lung’s microstructure produces an accumulation of inflammatory and immune cells in the interstitium and airspaces. Necrosis of type I cells results in denuded areas of the alveolar walls. Fluid and proteins leak into the alveolar spaces, forming fibrin-rich hyaline membranes. Following this exudative phase, processes begin that lead to either repair or further injury and ultimately fibrosis. Although the events that produce injury to the alveolar epithelial cells are unknown, many theories exist. As with many other diseases, there is a viral theory attempting to explain the repetitive damaging process, since DNA of the Epstein–Barr virus has been found in the lung tissue of patients with idiopathic pulmonary fibrosis (IPF). Human herpesviruses also are present, but their significance remains unknown. Gastroesophageal reflux disease (GERD) has also been associated with recurrent alveolar cell injury, as individuals with IPF have a high prevalence of GERD. Tobacco smoke has been associated, as well as an aberrant apoptotic response in the alveolar epithelial cells.
Fibrosis results from an increased number of fibroblasts, their passage into airspaces through damaged alveolar walls, and enhanced synthesis of extracellular matrix. Macrophages and lymphocytes stimulate fibroblast migration, proliferation, and synthetic activity. Alveolar epithelial cells, in addition to failing to function adequately in the reepithelization of the alveolar wall, may elaborate profibrotic cytokines. Peptides produced by matrix molecule degradation are chemotactic for fibroblasts. Fibronectin, which is present in the alveolar exudate and produced by activated macrophages, attracts fibroblasts and binds them to fibrin. Myofibroblasts have a phenotype intermediate between a fibroblast and a muscle cell and play an important role in wound healing. However, in the pathologic process of
lung fibrosis, they also provide the necessary scaffold over which definite areas of fibrosis will develop. They induce apoptosis in the hyperplastic pneumonocytes, impairing reepithelization of the denuded alveolar surface. Along with their expression of multiple profibrotic proteins, a reduction and alteration in the expression of different metalloproteinases is also observed. Metalloproteinases are the enzymes that normally degrade the excessive extracellular matrix deposited, including collagen fibers. Under normal reparative circumstances, the fibrotic process finishes with the apoptosis of the fibroblasts/myofibroblasts and degradation of the extracellular matrix. An imbalance between profibrotic substances and antifibrotic ones, with impairment in the secretion or abnormal synthesis of metalloproteinases, produces an excessive deposition of extracellular matrix, leading to the irreversible nature of the pulmonary fibrotic process.
lung fibrosis, they also provide the necessary scaffold over which definite areas of fibrosis will develop. They induce apoptosis in the hyperplastic pneumonocytes, impairing reepithelization of the denuded alveolar surface. Along with their expression of multiple profibrotic proteins, a reduction and alteration in the expression of different metalloproteinases is also observed. Metalloproteinases are the enzymes that normally degrade the excessive extracellular matrix deposited, including collagen fibers. Under normal reparative circumstances, the fibrotic process finishes with the apoptosis of the fibroblasts/myofibroblasts and degradation of the extracellular matrix. An imbalance between profibrotic substances and antifibrotic ones, with impairment in the secretion or abnormal synthesis of metalloproteinases, produces an excessive deposition of extracellular matrix, leading to the irreversible nature of the pulmonary fibrotic process.
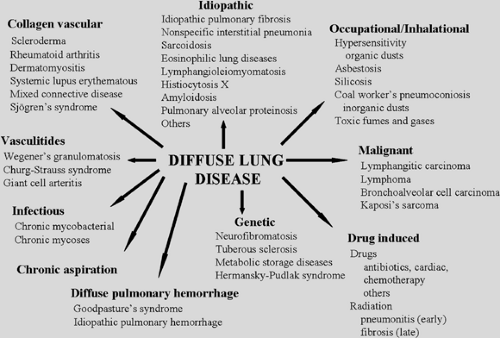 Figure 97-1. Causes of diffuse parenchymal lung disease. |
Table 97-1 Common Abbreviations for Diffuse Parenchymal Lung Diseases | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Organization of the exudate results in accumulation of fibrous tissue onto the alveolar wall. Type II pneumocytes proliferate, differentiate into type I cells, and epithelialize the surface of the scar. Widening of the normally thin air–blood interface is the end result.
The cells and mediators leading to fibrosis are part of the lung’s normal repair mechanisms. In reality, many injuries do not cause histologic or physiologic sequelae, the alveolar exudate is cleared, and the denuded alveolar walls undergo reepithelization without fibrosis. The outcome in any individual case is determined by the severity of the initial damage, the chronicity or repetition of the injury, the specific toxin or disease, dysregulation and autonomy of the inflammatory reaction, and individual susceptibility, including the patient’s age, with younger people having a better reparative capacity. However, the factors that determine uncomplicated resolution of injury versus progressive fibrosis remain incompletely understood. Reconsideration of mechanisms of fibrosis has led to the view that disordered healing in response to injury, and not persistent inflammation, may be the pivotal fibrogenic disturbance.97
Pathology
Although some DPLDs exhibit unique histopathologic features and in some cases microorganisms or inhaled irritants are identified, the findings are often nonspecific and reflect stereotyped patterns of response to injury. Individual patients’
pathology reports often describe diffuse alveolar damage, usual desquamative or nonspecific interstitial pneumonia, bronchiolitis obliterans–organizing pneumonia, or honeycombing. Figure 97-2 shows some of these most common histopathologic patterns seen in DPLD. However, most importantly, the clinician must recognize that in most cases the histopathologic findings alone are not sufficient to establish a specific clinical diagnosis. Indeed, the histopathologic findings can be common to several diagnostic possibilities, and correlation of the clinical history, physical examination, and laboratory and radiographic findings is necessary to establish the clinical diagnosis. A dynamic process involving close communication between the clinician, radiologist, and—in cases where histopathologic specimens have been obtained—the pathologist is required for the specific diagnosis of DPLDs. Described below are the histopathologic patterns commonly found in DPLD.
pathology reports often describe diffuse alveolar damage, usual desquamative or nonspecific interstitial pneumonia, bronchiolitis obliterans–organizing pneumonia, or honeycombing. Figure 97-2 shows some of these most common histopathologic patterns seen in DPLD. However, most importantly, the clinician must recognize that in most cases the histopathologic findings alone are not sufficient to establish a specific clinical diagnosis. Indeed, the histopathologic findings can be common to several diagnostic possibilities, and correlation of the clinical history, physical examination, and laboratory and radiographic findings is necessary to establish the clinical diagnosis. A dynamic process involving close communication between the clinician, radiologist, and—in cases where histopathologic specimens have been obtained—the pathologist is required for the specific diagnosis of DPLDs. Described below are the histopathologic patterns commonly found in DPLD.
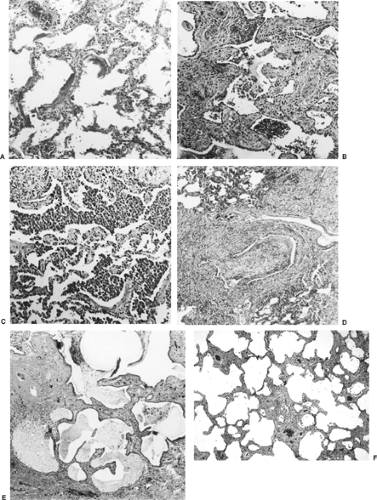 Figure 97-2. Common histopathologic patterns in diffuse lung disease. A: Diffuse alveolar hemorrhage (DAD). Alveolar cells are replaced by hyaline membranes. Inflammatory cell infiltrates are sparse at this early stage of injury. B: Usual interstitial pneumonia (UIP). Features of patchy chronic inflammation include fibrosis and interstitial cellular infiltrates, honeycombing, and reactive pneumocytes. C: Desquamative interstitial pneumonia (DIP). Alveoli with minimal architectural distortion are filled with macrophages. Pneumocyte hyperplasia is apparent. D: Cryptogenic organizing pneumonia (COP). An inflamed bronchiole is filled with a plug of granulation tissue. Chronic active inflammation persists around the airway. E: Honeycomb lung. Normal parenchyma is replaced by fibrosis and epithelium-lined cystic changes. Cysts are filled with mucus and inflammatory cells. F: Nonspecific interstitial pneumonia (NSIP). An inactive homogeneous linear pattern of alveolar septal fibrosis with some preservation of the alveolar architecture. |
Diffuse alveolar damage (DAD) (Fig. 97-2A) is typical of severe acute injury. Katzenstein and associates43 introduced the term to describe the features of adult respiratory distress syndrome (ARDS). Early cases show interstitial and alveolar edema, epithelial necrosis, and sloughing, leading to denuded alveolar walls, acute inflammatory cells, and hyaline membranes. Later, there is organization and repair (proliferative phase), with proliferation of fibroblasts in the interstitium and airspaces
and of type II pneumocytes along the damaged alveolar walls, along with partial or complete resorption of the hyaline membranes seen earlier. The proliferative phase is thought to occur within 1 to 2 weeks after the initial injury. DAD is generally found diffusely throughout the parenchyma and is histologically uniform. These hallmarks suggest a discrete generalized injury. In addition to ARDS, DAD may be seen in other acute syndromes, including acute interstitial pneumonia (Hamman–Rich syndrome), acute radiation pneumonitis, drug reactions, inhalation injury, hypersensitivity pneumonitis, and acute lupus pneumonitis. DAD in immunocompromised patients is often due to infection. The term acute interstitial pneumonitis has been suggested by Katzenstein and colleagues44 to describe idiopathic cases, especially to distinguish this lesion from the more common chronic interstitial pneumonias.
and of type II pneumocytes along the damaged alveolar walls, along with partial or complete resorption of the hyaline membranes seen earlier. The proliferative phase is thought to occur within 1 to 2 weeks after the initial injury. DAD is generally found diffusely throughout the parenchyma and is histologically uniform. These hallmarks suggest a discrete generalized injury. In addition to ARDS, DAD may be seen in other acute syndromes, including acute interstitial pneumonia (Hamman–Rich syndrome), acute radiation pneumonitis, drug reactions, inhalation injury, hypersensitivity pneumonitis, and acute lupus pneumonitis. DAD in immunocompromised patients is often due to infection. The term acute interstitial pneumonitis has been suggested by Katzenstein and colleagues44 to describe idiopathic cases, especially to distinguish this lesion from the more common chronic interstitial pneumonias.
Liebow and Carrington54,55 have described the classic interstitial pneumonias. They described five types of chronic interstitial pneumonia: usual interstitial pneumonia (UIP), bronchiolitis obliterans interstitial pneumonia and diffuse alveolar hemorrhage (BIP), desquamative interstitial pneumonia (DIP), lymphocytic interstitial pneumonia (LIP), and giant-cell interstitial pneumonia (GIP). Most frequently encountered is UIP (Fig. 97-2B). Like DAD, UIP represents a nonspecific response. The clinical syndrome, however, typically evolves over a longer period of time. Hyaline membranes, edema, and alveolar exudates are rarely seen. The distribution is patchy and the histology is nonuniform, varying from normal alveoli to inflammation to fibrosis. This pattern suggests repetitive or low-grade chronic injury. Areas of UIP may be seen in idiopathic pulmonary fibrosis (IPF), collagen vascular disease, chronic hypersensitivity pneumonitis, some of the pneumoconioses, sarcoidosis, drug-induced DPLD, granulomatous infections, late radiation pneumonitis, healed infections, organized DAD, chronic aspiration, histiocytosis X, and even in cases of chronic pulmonary edema due to passive congestion. In desquamative interstitial pneumonia (DIP) (Fig. 97-2C), the involvement is diffuse and the histology homogeneous. It was initially believed that the highly cellular histology reflected desquamated alveolar epithelial cells, but it is now known that filling of the alveoli by macrophages is the defining feature. Interstitial monocytic infiltration is also seen, but fibrosis is minimal. A DIP pattern may be found in idiopathic DIP, histiocytosis X, drug reactions (e.g., amiodarone toxicity), alveolar hemorrhage, pneumoconioses, eosinophilic pneumonias, obstructive pneumonia (in which case the lesion is not diffuse), lipid pneumonia and lipid storage diseases, respiratory bronchiolitis, and infections in the immunosuppressed patient. In Europe, UIP and DIP have been termed mural and desquamative fibrosing alveolitis, respectively.
Carrington and associates11 viewed UIP and DIP as two diseases because of different pathologic findings and because DIP is more amenable to treatment. Others, however, agree that DIP is the early cellular phase and UIP the later mixed or fibrotic phase of the same reaction.78,94,107 As noted, there is much overlap in clinical processes in which the two patterns may be identified. In addition, areas of DIP can be found in cases of predominant UIP histology, and DIP has been shown to progress to UIP. Recent observations of the frequent association with cigarette smoking and pathologic similarities have linked the condition with the spectrum of respiratory bronchiolitis.109
Lymphocytic interstitial pneumonia (LIP) consists of a dense interstitial infiltrate of lymphocytes, plasma cells, and macrophages. The alveolar septa are typically extensively infiltrated. Granulomas, lymphoid follicles, and amyloid deposits are often present. Although an idiopathic form exists, LIP has a strong association with immunologic diseases, acquired immunodeficiency syndrome (AIDS), and lymphoid malignancies. For these reasons LIP is now classified by some as a lymphoproliferative disorder rather than an idiopathic interstitial pneumonia.
Its histologic pattern is, however, unequivocally that of an interstitial pneumonia, and it thus has remained in the current classification of idiopathic interstitial pneumonias of the American Thoracic Society (ATS)/European Respiratory Society (ERS).105 Giant-cell interstitial pneumonia (GIP) is a rare pattern in which bizarre multinucleated cells fill the airspaces. Most cases result from inhalation of hard metal particles; thus it has been dropped from its former classification within the category of idiopathic interstitial pneumonias.73
Its histologic pattern is, however, unequivocally that of an interstitial pneumonia, and it thus has remained in the current classification of idiopathic interstitial pneumonias of the American Thoracic Society (ATS)/European Respiratory Society (ERS).105 Giant-cell interstitial pneumonia (GIP) is a rare pattern in which bizarre multinucleated cells fill the airspaces. Most cases result from inhalation of hard metal particles; thus it has been dropped from its former classification within the category of idiopathic interstitial pneumonias.73
Davison and colleagues21 described cryptogenic organizing pneumonitis (COP), and shortly thereafter Epler and colleagues25 described the same entity under the name bronchiolitis obliterans organizing pneumonia (BOOP) (Fig. 97-2D). BOOP became the more commonplace terminology. However, the term cryptogenic organizing pneumonitis (COP) is preferable, since it avoids confusing other similarly named but separate and distinct airway diseases such as constrictive bronchiolitis obliterans with BOOP. In COP, organizing connective tissue partially or totally fills alveoli, respiratory bronchioles, and alveolar ducts. Bronchiolar intraluminal polypoid plugs may or may not be seen. Mononuclear cell infiltrates are seen in the airway walls, and the alveoli often contain lipid-filled macrophages. Fibrosis is absent or minimal. The anatomic distribution is patchy but histologically uniform. Background lung architecture is relatively preserved. Like other nonspecific reactions, COP can be idiopathic or associated with the response to other forms of lung injury, such as organizing infection, drug reactions, inhalation injury, collagen vascular disease, hypersensitivity pneumonitis, and chronic eosinophilic pneumonia.
Progressive severe fibrosis results in cyst formation and loss of normal architecture. When this pattern coexists with normal and inflamed areas, as in UIP, it is called “honeycombing” (Fig. 97-2E). An etiologic diagnosis can often be made if the histology of the less fibrotic areas is characteristic. “Honeycomb lung” denotes diffuse distortion and represents the potential end stage of any fibrogenic process; in practice, however, it is seen mainly in IPF, histiocytosis X, scleroderma, rheumatoid lung, asbestosis, LAM, and sarcoidosis.
Katzenenstein and Fiorelli45 detailed the features of an additional variant of the interstitial pneumonias of unknown etiology entitled nonspecific interstitial pneumonia (NSIP) (Fig. 97-2F). Although the characteristic lesion contains varying proportions of interstitial inflammation and fibrosis, NSIP differs from UIP in that all areas appear to be temporally uniform. In addition to an idiopathic form, NSIP also occurs in AIDS patients and others who are immunocompromised as well as in patients with collagen vascular disease, inhalation of organic antigens, and resolving acute lung injury. In a report of 12 cases, 6 were idiopathic, 3 were associated with collagen vascular disease (CVD), 2 were associated with organic dust exposure, and 1 followed an episode of ARDS.18 The response to treatment is better than for UIP but appears to be more variable than in COP.
In some DPLDs, necrotizing or nonnecrotizing granulomas are a universal or occasional feature. Processes associated with granuloma formation include sarcoidosis, granulomatous infections, talcosis, berylliosis, Wegener’s granulomatosis, necrotizing sarcoid granulomatosis, bronchocentric granulomatosis, certain drug reactions, and hypersensitivity reactions.
A recent international consensus document provides updated standards for the classification of idiopathic DPLD—specifically idiopathic pulmonary fibrosis (IPF), also known as cryptogenic fibrosing alveolitis (CFA). This is detailed in a communication by the ATS and the ERS.105 The current ATS/ERS classification for idiopathic interstitial pneumonia recognizes the following clinicopathologic entities in order of relative frequency: idiopathic pulmonary fibrosis/cryptogenic fibrosing alveolitis, nonspecific interstitial pneumonia, cryptogenic organizing pneumonia, acute interstitial pneumonia, respiratory bronchiolitis–associated interstitial lung disease, desquamative interstitial pneumonia, and lymphoid interstitial pneumonia.
Epidemiology
The true incidence of DPLD is unknown. Coultas and colleagues,19 from a population-based registry of patients with DPLDs in Bernalillo County, New Mexico, found an incidence of 30 per 100,000 per year, with almost one-third of the cases being idiopathic pulmonary fibrosis. The incidence was higher for men than for women. The estimated prevalence was almost three times as high as the incidence, implying a mean survival time of almost 3 years. Table 97-2 gives the relative proportions of the causes of DPLD. Many of these usually described in textbooks are uncommon in clinical practice; in a typical internal medicine practice, three-fourths of the cases represent one of three diagnoses: idiopathic pulmonary fibrosis (IPF) (also called usual interstitial pneumonia [UIP]), sarcoidosis, or collagen vascular disease–associated interstitial lung disease (especially nonspecific interstitial pneumonia [NSIP]).2
DPLDs demonstrate specific age preferences. DPLDs occurring in infancy and childhood include follicular bronchitis, cellular interstitial pneumonia, and acute idiopathic pulmonary hemorrhage of infancy. Following adolecscence and prior to age
40, familial IPF, metabolic storage disorders, Hermansky–Pudlak syndrome, and other inherited DPLDs should be considered. Sarcoidosis tends to occur in young to middle-aged people but can present at any age. IPF most commonly occurs after age 50.70
40, familial IPF, metabolic storage disorders, Hermansky–Pudlak syndrome, and other inherited DPLDs should be considered. Sarcoidosis tends to occur in young to middle-aged people but can present at any age. IPF most commonly occurs after age 50.70
| ||||||||||||||||||||||||||||||||
Genetic predispositions for DPLDs exist. LAM and tuberous sclerosis–associated ILD occur almost exclusively in women. Because of their increased risk for autoimmune diseases, women are more likely to have ILD associated with collagen–vascular disease. Men are at greater risk for pneumoconiosis. Finally, African Americans have a 10 to 12 times higher incidence of sarcoidosis than whites.70
Clinical Approach to Patients with DPLD
Clinical History
The clinical course for patients with DPLD may be acute or prolonged. It may be indolent over many years or rapidly progress to life-threatening respiratory failure and death. A differential diagnosis can often be created by carefully focusing on the patient’s medical history, including the duration of symptoms, medications, tobacco usage, and environmental and occupational exposures, including hobbies, pets, and travel history. Antecedent diseases such as prior malignancies, drug regimens, exposure to dusts or animals, and risk factors for AIDS are examples of information that may suggest a diagnosis.
Respiratory bronchiolitis-associated ILD, desquamative interstitial pneumonia, and Langerhans cell histiocytosis X occur almost exclusively in patients with a smoking history. Although a smoking history is commonplace in patients with IPF, it is less common in patients with hypersensitivity pneumonitis and sarcoidosis.
Occupational exposures, if elicited in the history, may obviate the need for invasive diagnostic testing. Silicosis is the most prevalent worldwide occupational DPLD, with exposure to crystalline silica occurring in brickyards, foundries, mines, and sandblasting sites. Table 97-3 lists commonly known exposures and their relation to DPLD.
A comprehensive review of the patient’s current and past medications may uncover a cause of DPLD. The list of medications associated with DPLD continues to grow. Chemotherapeutic agents—including bleomycin, methotrexate, and mitomycin C—are known to cause DPLD. Other commonly associated drugs include amiodarone, nitrofurantoin, nonsteroidal anti-inflammatory agents, diphenylhydantoin, and penicillamine. Intravenous drug abuse can cause an inflammatory reaction related to particulate matter intermixed with the drug, resulting in a pulmonary foreign body granulomatosis. Radiation therapy can also result in both acute and chronic forms of DPLD.
Patients with DPLD may have a genetic predisposition. A history of DPLD among first-degree relatives raises the possibility of a hereditary DPLD, such as familial pulmonary fibrosis.84 Additionally, DPLD is more commonplace in patients with the inherited diseases of neurofibromatosis, tuberous sclerosis, Hermansky–Pudlak syndrome, and metabolic storage disorders.84
Most patients come to medical attention because of shortness of breath. Although dyspnea associated with infections or hypersensitivity pneumonitis may come on abruptly and progress rapidly, the clinical syndrome is subacute or chronic in most processes. In pneumoconiosis, dyspnea may not only develop insidiously but also appear many years following exposure. Acute symptoms rapidly progressing to respiratory failure suggest a diagnosis of acute interstitial lung disease or acute eosinophilic pneumonia. Table 97-4 outlines DPLDs and their relation to symptom duration.
Table 97-3 Association of Selected Environmental Exposures and Diffuse Parenchymal Lung Disease (DPLD) | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
Although cough is a nonspecific finding, a nonproductive cough is common in both acute and chronic DPLDs. Cough can be associated with respiratory bronchiolitis-associated interstitial lung disease, sarcoidosis, hypersensitivity pneumonitis, and GERD. In fact, GERD is a common cause of chronic cough, and chronic aspiration should be considered in patients with cough and DPLD.34 Lymphangitic carcinomatosis can cause a chronic cough. With bronchoalveolar cell carcinoma, both cough and bronchorrhea can be seen. In long-standing DPLD, cough can become productive and refractory to conventional treatments. Hemoptysis is unusual in most DPLDs but can occur with diffuse pulmonary hemorrhage, Goodpasture’s syndrome, Wegener’s granulomatosis, and LAM. New-onset hemoptysis in patients with known DPLD should not reflexively be attributed to the DPLD but instead may be related to underlying infection, pulmonary embolus, or malignancy.
Fever, weight loss, fatigue, and myalgias can occur in infectious and noninfectious disorders. The presence of these symptoms suggests an underlying collagen vascular disease or more acute DPLD, such as acute interstitial pneumonia or BOOP/COP. Patients with sarcoid commonly have fever and other systemic symptoms. Some forms of hypersensitivity
pneumonitis, such as farmer’s lung, can present with fever, chills, and dyspnea.
pneumonitis, such as farmer’s lung, can present with fever, chills, and dyspnea.
Table 97-4 Diffuse Parenchymal Lung Diseases and Symptom Duration | ||
|---|---|---|
|
Chest pain can occur commonly in systemic lupus erythematosus and occasionally in mixed connective tissue disease, Wegener’s granulomatosis, and rheumatoid arthritis. Sarcoidosis can present with vague chest discomfort or significant pleuritic chest pain. Pleuritic chest pain can also occur with pneumothorax, which can be secondary to LAM, tuberous sclerosis, Langerhans cell histiocytosis, and neurofibromatosis.
Other nonpulmonary symptoms may be more specific, such as the combination of uveitis, parotiditis, and erythema nodosum in sarcoidosis, known as Heerfordt’s syndrome, and sinusitis–otitis in Wegener’s granulomatosis. Arthritic symptoms may be seen in sarcoidosis and connective tissue–induced DPLD. Spontaneous pneumothorax may be the presenting event in LAM and Langerhans cell histiocytosis. GERD may be associated with aspiration pneumonia as well as scleroderma and mixed connective tissue diseases. Neurologic symptoms may reflect a vasculitis, sarcoidosis, or tuberous sclerosis.
Physical Examination
Breathlessness and tachypnea may be apparent at rest or provoked by exertion. Javaheri and Sicilian39 demonstrated that ventilation is maintained and hypercapnia avoided by rapid shallow breathing. The physical examination may be normal and the lung exam nonspecific. Fine crackles reminiscent of the sound of Velcro or cellophane are the classic finding not only in IPF but also other DPLDs. The crackles, when present, continue throughout inspiration and are predominantly basilar in location. With disease progression, the crackles may progress toward the lung apices. Patients with sarcoidosis and other granulomatous DPLDs commonly do not have respiratory crackles. Midinspiratory squeaks occur in DPLD with airway-centered pathology, such as hypersensivity pneumonitis, BOOP/COP, and constrictive bronchiolitis. Squeaks may also occur with NSIP. Wheezing can occur in a subset of DPLDs, such as sarcoidosis, Langerhans cell histiocytosis, and LAM.
Signs of secondary pulmonary hypertension may be encountered in advanced DPLD and include jugular venous distention, increased intensity of the pulmonic second heart sound, tricuspid regurgitation, and lower extremity edema. Hepatomegaly and ascites may also be found.
Digital clubbing is most common in patients with respiratory diseases. Clubbing can be seen in 25% to 50% of patients with IPF, desquamative interstitial pneumonia, and chronic hypersensitivity pneumonitis, and in 75% of patients with DPLD related to rheumatoid arthritis. Clubbing is rare in other types of connective tissue disease–induced DPLD as well as in patients with sarcoidosis, respiratory bronchiolitis–associated interstitial lung disease, BOOP/COP, and other acute and chronic DPLDs.
Many of the DPLDs exhibit skin abnormalities. Some 25% of patients with sarcoidosis have cutaneous involvement, with the classic findings being erythema nodosum and lupus pernio. Subcutaneous nodules, particularly in the vicinity of joints, can occur with rheumatoid arthritis. Various skin abnormalities are commonplace with the connective tissue diseases: scleroderma, systemic lupus erythematosus, and dermatomyositis as well as neurofibromatosis, tuberous sclerosis, amyloidosis, and pulmonary Langerhans cell histiocytosis.
Laboratory Testing
Laboratory tests alone do not confirm a specific diagnosis but may be supportive in the proper clinical setting. Routine tests include a complete blood count (CBC) with differential, platelet count, and blood chemistries including calcium and liver function tests. For suspected hypersensitivity pneumonitis, serum precipitins focused on known exposures may be performed, but both false-negatives and the results of prior sensitization may limit their usefulness. An elevated level of angiotensin converting enzyme (ACE) is neither specific nor sensitive enough to establish a diagnosis of sarcoidosis. Increased ACE levels may be seen in many other granulomatous lung diseases. Nonetheless, when the ACE level is elevated in sarcoidosis, it can serve as a marker of disease activity and thus be helpful in ongoing follow-up. When connective tissue diseases are suspected, anti- nuclear antibody (ANA) and rheumatoid factor are checked. For pulmonary vasculitides, cytoplasmic antineutrophil cytoplasmic antibody (C-ANCA), perinuclear antineutrophil cytoplasmic antibody (P-ANCA), antiglomerular basement membrane antibody (anti-GBM antibody), ANA, and urine sediment should be checked. A positive C-ANCA is most suggestive of Wegener’s granulomatosis, with an overall estimated sensitivity of 81% and specificity of 98%. P-ANCA may be positive in Wegener’s granulomatosis as well as in microscopic polyangiitis, Churg–Strauss syndrome, and other vasculitides. A positive anti-GBM antibody
in a patient with diffuse alveolar hemorrhage is diagnostic of Goodpasture’s syndrome.
in a patient with diffuse alveolar hemorrhage is diagnostic of Goodpasture’s syndrome.
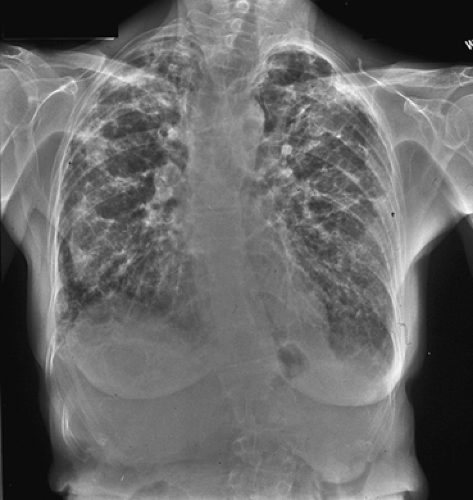 Figure 97-3. Chest radiograph with diffuse bilateral infiltrates of idiopathic pulmonary fibrosis (IPF). There are net-like linear and curvilinear (i.e., reticular) densities, with a slight lower lobe predominance noted here. |
Radiographic Features
A PA and lateral chest x-ray (CXR) is the initial radiographic study for almost all patients with respiratory symptoms. A diffusely abnormal CXR typically showing bilateral infiltrates (Fig. 97-3) may often be the first finding alerting the physician to possible DPLD. Although the CXR may be entirely normal in 10% of patients with DPLD, the CXR continues to be the basic imaging tool for the initial evaluation and subsequent DPLD surveillance,24 The radiographic distribution of the CXR abnormalities can help in narrowing the differential diagnosis. Efforts should be made to obtain old CXRs, which may allow for determination of the DPLD’s onset, chronicity, stability, or rate of progression. In general, DPLD produces four patterns: reticular, nodular, reticulonodular, and linear.
Reticular infiltrates appear as a network of curvilinear opacities and are described as fine, medium, or coarse, depending on the size of the mesh (see Fig. 97-2). These gradations generally correlate with the progression from early alveolitis to advanced fibrosis. Transition over time from fine to medium to coarse patterns has been documented for IPF and rheumatoid lung.57,113 Coarse reticulation (thick-walled cystic spaces at least 5 mm in diameter) indicates severe fibrosis and, like its pathologic counterpart, is termed honeycombing. Nodular opacities consist of well-circumscribed, small, round opacities of varying diameter. More common is a reticulonodular pattern that can result from superimposition of nodules and reticulation or from an end-on orientation of reticular opacities in the absence of histologic nodules. Thickening of the interlobular septa and perivascular sheaths produces a linear pattern typified by Kerley B lines. Early acinar opacities may simulate nodularity, but the borders are indistinct. Coalescence produces airspace consolidation. Patent airways surrounded by opacified acini appear as air bronchograms. Mixed interstitial and airspace patterns are common.
Ground-glass opacification results from changes below the resolution of the imaging technique, including fine reticulation, micronodularity, and partial filling of the alveolar spaces or thickening of the interstitium. For standard CXRs, this description refers to a generalized, sometimes granular haziness. On computed tomography (CT), ground-glass appearance is often patchy and is defined as increase in lung density that does not obscure bronchovascular structures.
Table 97-5 lists other features of the CXR—including anatomic distribution, overall lung volume, and extrapulmonary findings—which may aid in diagnosis.
The limitations of conventional CXRs in DPLD must be kept in mind and include limited sensitivity, an incomplete view of the lungs and mediastinum, variability in interpretation, and a lack of specificity of the aforementioned plain film patterns. On CXR, as much as 40% of the lung parenchyma is obscured by superimposed structures. The frequent inadequacy of CXR was highlighted by McLoud64 and colleagues, who interpreted films from 365 biopsy-confirmed cases of DPLD and, using a standardized system, recorded the three most likely diagnoses. Despite a correlation between certain diseases and patterns, the correct diagnosis was included in the first two choices only 50% of the time.
Although conventional CT is better than conventional CXR, it is still relatively insensitive in the imaging of DPLD. High-resolution CT (HRCT) is currently a critical imaging tool in the assessment of DPLD. In addition to the benefits of conventional CT—including the visualization of all lung regions, identification of mediastinal and hilar lymphadenopathy, and small effusions—HRCT allows superior imaging of the lung and its airways. HRCT gives 1- to 1.5-mm-thick slices at 10-mm intervals in order to obtain better lung parenchymal detail.
The resolution of HRCT reliably extends to the anatomic level of the secondary pulmonary lobule. Colby and Swenson14 have provided a comprehensive analysis of the HRCT correlates of the histopathologic patterns in DPLD. The value of HRCT was also confirmed by Mathiesen and colleagues,63 who were able to make a correct first-choice diagnosis in 76% of cases, compared with 57% by CXR alone. Because of characteristic CT patterns, high accuracy was achieved in silicosis (93%) (Fig. 97-4), UIP-IPF (89%) (Fig. 97-5), lymphangitic carcinoma (85%) (Fig. 97-6), histiocytosis X (83%) (Fig. 97-7), and sarcoidosis (77%) (Fig. 97-8). Strollo101 has provided a valuable review of HRCT in DPLD. As with plain CXR, however, a normal HRCT does not eliminate the possibility of DPLD. Orens and colleagues74 found that 12% of dyspneic patients had normal HRCT findings, despite measured impairment of gas exchange and biopsy-proven DPLD, although the patients with false-negative results had less severe functional and histologic findings than those with abnormal HRCT findings. Although HRCT may be nonspecific and sometimes falsely negative, HRCT imaging often narrows the list of differential diagnoses at presentation, and the combination of clinical–radiographic correlation may be diagnostic in many instances (e.g., idiopathic pulmonary fibrosis, sarcoidosis, lymphangitic carcinomatosis, and certain forms of hypersensitivity
pneumonitis). Several DPLDs demonstrate classic findings that are highly suggestive of a diagnosis, including pulmonary alveolar proteinosis (Fig. 97-9), LAM (Fig. 97-10), and Langerhans cell histiocytosis. Table 97-6 summarizes useful CT patterns that may help in establishing a DPLD differential diagnosis. HRCT in conjunction with the clinical history, physical exam, blood tests, and pulmonary function tests (PFTs) may obviate the need for more invasive diagnostic procedures. The HRCT appearance may also be valuable in determining disease activity and response to treatment, especially in IPF. Ground-glass patterns suggest an improved response to treatment and survival.
pneumonitis). Several DPLDs demonstrate classic findings that are highly suggestive of a diagnosis, including pulmonary alveolar proteinosis (Fig. 97-9), LAM (Fig. 97-10), and Langerhans cell histiocytosis. Table 97-6 summarizes useful CT patterns that may help in establishing a DPLD differential diagnosis. HRCT in conjunction with the clinical history, physical exam, blood tests, and pulmonary function tests (PFTs) may obviate the need for more invasive diagnostic procedures. The HRCT appearance may also be valuable in determining disease activity and response to treatment, especially in IPF. Ground-glass patterns suggest an improved response to treatment and survival.
Table 97-5 Features of Diffuse Parenchymal Lung Diseases on Routine Chest Radiography | ||
|---|---|---|
|
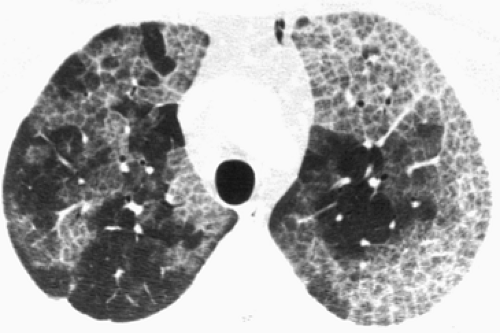 Figure 97-4. Acute silicosis in a sandblaster. HRCT demonstrates patchy ground-glass opacity and interlobular thickening typical of acute silicosis. (From Webb WR, Müller NL, Naidich DP. Pneumoconiosis, occupational, and environmental lung disease. In Webb WR, Müller NL, Naidich DP. High Resolution CT of the Lung, 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2009:318. With permission.) |
Pulmonary Function Testing
PFTs should include spirometry, lung volumes, and diffusion capacity (DLCO). PFTs generally demonstrate a restrictive pattern exhibiting symmetrically decreased lung volumes with varying degrees of gas exchange abnormalities and hypoxemia. In a symptomatic patient with radiographic evidence of DPLD and normal PFTs, the only abnormality may be oxygen desaturation
with exercise. Thus a pulmonary gas exchange test, both at rest and with exercise, utilizing pulse oximetry should be obtained. The 6-minute walk test is gaining popularity in the evaluation of patients with DPLD as well as in assessing their response to treatment.
with exercise. Thus a pulmonary gas exchange test, both at rest and with exercise, utilizing pulse oximetry should be obtained. The 6-minute walk test is gaining popularity in the evaluation of patients with DPLD as well as in assessing their response to treatment.
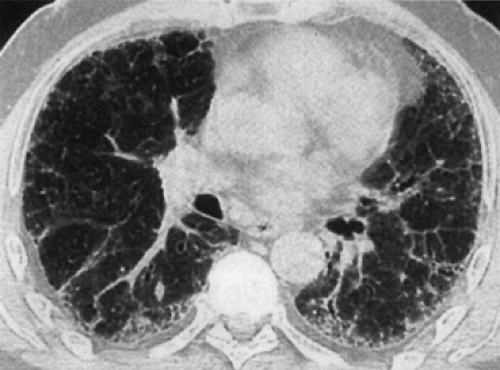 Figure 97-5. Typical usual interstitial pneumonia (UIP). HRCT showing the characteristic findings of a reticular infiltrative process, architectural distortion, and honeycombing in a peripheral distribution. UIP is usually more prominent in the lung bases. In later stages of UIP, there are cicatricial changes that may result in traction bronchiectasis and bronchiolectasis. (From Stern EJ, Swensen SJ. Fibrotic lung diseases. In Stern EJ, Swensen SJ. High Resolution CT of the Lung, 2nd ed. Philadelphia: Lippincott Williams & Wilkins, 2001:190. With permission.) |
Although PFTs cannot diagnose a specific DPLD, they can help in the differential diagnosis and are important in the objective assessment of respiratory symptoms. In the early stages of DPLD, the DLCO may be the only abnormality and is a sensitive though nonspecific measurement of disease severity. DLCO may be decreased out of proportion to other portions of the PFTs, indicating concomitant pulmonary vascular disease. This may also be seen occasionally in LAM, Langerhans cell histiocytosis, and pulmonary alveolar proteinosis.
An obstructive component to the PFTs may be found in some cases, which can aid in limiting the differential diagnosis. Specifically, causes of DPLD that demonstrate a mixed pattern of obstruction and restriction include eosinophilic pneumonia, hypersensitivity pneumonitis, Langerhans cell histoplasmosis, LAM, respiratory bronchiolitis–interstitial lung disease, and sarcoidosis. In cryptogenic organizing pneumonia (COP) and constrictive bronchiolitis, an obstructive pattern without restriction may be seen.
Respiratory muscle weakness, which can be seen in polymyositis, scleroderma, or systemic lupus erythematosus, may show decreased maximal inspiratory and expiratory pressures and maximum minute ventilation.
Diagnostic Approach
A synthesis of the history, physical examination, laboratory data, and radiographs may be diagnostic. Invasive diagnostic procedures are warranted when the clinical synthesis does not yield a confident diagnosis, the clinical course is atypical, or on occasion when it is necessary to assess disease activity. Although biopsy of extrathoracic sites, percutaneous aspiration of the lung or pleura, thoracentesis, or mediastinoscopy may be appropriate in some cases, the primary diagnostic methods for DPLD are bronchoalveolar lavage (BAL), transbronchial lung biopsy (TBLBx), and surgical biopsy. The technical aspects of these procedures are described elsewhere in this text. Figure 97-11 is an algorithm for diagnosing DPLD. The likelihood of establishing a specific diagnosis with this algorithm is greatest for hypersensitivity pneumonitis, Langerhans cell histoplasmosis, lymphangitic carcinoma, sarcoidosis, silicosis, and possibly IPF.
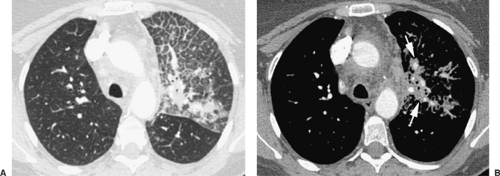 Figure 97-6. Lymphangitic carcinomatosis from breast cancer. A: Lung window image from a contrast-enhanced HRCT demonstrating extensive interlobular thickening and peribronchovascular thickening in the left upper lobe. B: Soft tissue window contrasts peribronchovascular tumor (arrows) and opacified pulmonary arteries. (From Webb WR, Müller NL, Naidich DP. Diffuse pulmonary neoplasms and pulmonary lymphoproliferative diseases. In Webb WR, Müller NL, Naidich DP. High Resolution CT of the Lung, 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2009:245. With permission.) |
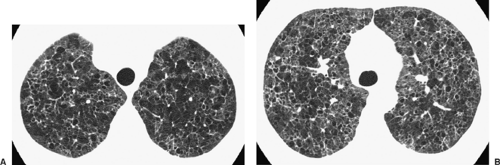 Figure 97-7. Langerhans cell histiocytosis. HRCT shows characteristic findings of a cystic infiltrative process with upper lobe predominance. The cysts can attain unusual shapes. (From Webb WR, Müller NL, Naidich DP. Diffuse pulmonary neoplasms and pulmonary lymphoproliferative diseases. In Webb WR, Müller NL, Naidich DP. High Resolution CT of the Lung, 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2009:136. With permission.) |
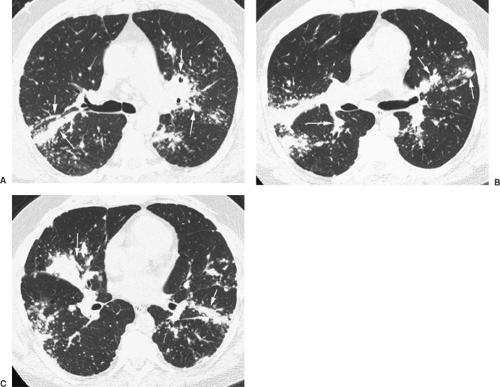 Figure 97-8. A-C: Sarcoidosis. HRCT showing extensive nodular involvement of the peribronchovascular interstitium (arrows) within both parahilar and peripheral lung. Small nodules are visible at the edges of larger masses, and extensive involvement of the pleural surfaces and fissures is also visible. (From Webb WR, Müller NL, Naidich DP. Diffuse pulmonary neoplasms and pulmonary lymphoproliferative diseases. In Webb WR, Müller NL, Naidich DP. High Resolution CT of the Lung, 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2009:276. With permission.) |
Table 97-6 Common High-Resolution CT Findings of Diffuse Parenchymal Lung Disease | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


