Diagnostic Decision Making Using Modern Technology: Introduction
The history of medicine has been marked by a perpetual contest, waged by those who propose innovative clinical techniques or technologies and those who resist its widespread use or adoption. As technologic advances in diagnostic imaging continue to leap forward, the cardiologist in the 21st century confronts this back and forth on a regular basis, with both sides offering salient arguments in their favor. History can offer insights into the potential risks and benefits of the implementation of technology and perhaps can provide a navigational guide to the current array of diagnostic testing available to the modern cardiologist.
Until the 19th century, physicians, while having a more precise understanding of the nature of disease in the deceased human body, still had considerable difficulty understanding and diagnosing diseases in the living. The gradual development and enthusiastic reception of technologic aids, then as now, represented remarkable advancements in the evolution of medical practice over the course of the last 200 years. Prior to the development of medical instrumentation, physicians were purely reliant on the patient’s subjective symptoms and a few signs that were readily observable. In 1761, Joseph Leopold Auenbrugger of Vienna published a groundbreaking work entitled Inventum Novum, in which he described a new diagnostic method he coined chest percussion. In his work, he documents how by using his technique, one could distinguish between sounds of a healthy chest and a chest affected by pneumonia or tuberculosis.1
Although Auenbrugger’s work enabled physicians to make more precise diagnoses, it was largely ignored for several years.2 Despite its limited implementation, it did indirectly influence a French physician named René Théophile-Hyacinthe Laennec. In 1816, drawing from his mentor’s enthusiastic acceptance of Auenbrugger’s technique, Laennec invented the stethoscope.3 In On Mediate Auscultation, Laennec describes the circumstances that led to his inspiration to use a cylindrical cone to listen to all manner of sounds in the chest. Like any new technologic innovation, the stethoscope had its detractors. Many physicians felt that using the stethoscope was undignified and ludicrous. However, Laennec, undeterred, meticulously correlated the sounds he heard through the stethoscope with postmortem pathologic findings. For many years after his initial description, even after its merits were becoming widely accepted, there was still considerable controversy regarding the technical and acoustic merits of monaural versus biaural instruments.2 Roughly 150 years later in the 1950s, Carl Hellmuth Hertz, a physicist, and Inge Elder, a cardiologist, were also using sound waves in an equally revolutionary way. By reflecting sound waves off of the heart, they were taking the first steps toward modern echocardiography.
It seems laughable today to question the merits of echocardiography or even the stethoscope, but in their day, each of these advances had their detractors and naysayers. The continual questioning and examination of the merits of technologic advances, however, is a vital part of their development. Although this Darwinian approach has seen many evolutionary leaps in diagnostic testing that have often led to improved outcomes for the sick and injured, a far greater number of tests have not withstood the klieg lights of scientific scrutiny. History is replete with a vast number of new technologies or techniques hailed initially as “breakthroughs“ or “revolutionary“ that ultimately fail to demonstrate improved outcomes in high-quality comparative trials. In the modern era, the alarming increases in health care costs associated with technologic advancement have drawn the attention of the government, the health care industry, and the public. In particular, diagnostic medical imaging, experiencing unprecedented growth, accounts for a large percentage of this increase in health care expenditures. The practicing cardiologist thus has a social responsibility to approach the adoption of new technologies and the utilization of imaging tests with a critical eye.4 The goal of this chapter is to help develop a framework that will help the modern physician understand when and how a new technology or technologic application should be adopted.
Fryback and Thornbury5 proposed that there were six basic conditions that needed to be met for a new diagnostic test to demonstrate clinical utility:
The equipment or technology has to meet industry and government standards of quality, safety, and reliability.
The test has to demonstrate accuracy in establishing or excluding the disease or condition for which it is applied.
The use of the test has to demonstrate a diagnostic impact; in other words, it has to achieve a diagnostic yield that exceeds the one expected without the use of the test.
The use of the test has to achieve a therapeutic impact, meaning that if the diagnosis is established, a specific treatment can be implemented.
The use of the test has to lead to improvement in patient’s outcomes.
The use of the test has to provide health and/or economic benefits to the society.
Approval of New Technology for Clinical Diagnostic Use
The origin and evolution of technology can be roughly hewed into two main forms of development. Akin to the theory of punctuated equilibrium in evolutionary biology, one model of technologic advancement involves a “breakthrough“ device or invention that represents a true paradigm shift. In human history and medical history, many possible examples of these events come easily to mind, including Thomas Edison’s light bulb, Wilhelm Roentgen’s accidental discovery of x-rays, and Laennec’s invention of the stethoscope. However, upon closer inspection, true leaps in discovery are exceedingly rare. Most advances in medical technology have been built on the back of work done by preceding generations or have drawn their inspiration from other disciplines. This second model of development, analogous to the theory of phyletic gradualism in evolutionary biology, acknowledges that advances are typically the result of a succession of relatively modest changes and refinements in existing technologies. For example, newer power sources, materials, components, or designs, although small, accumulate over time, transforming a medical device and affecting its clinical effectiveness. In this way, seemingly small changes can amass, bringing about a generational change. These changes can lead to new uses and improved outcomes for patients. The development of imaging procedures such as computed tomography (CT) and positron emission tomography, and now hybrid positron emission tomography/CT, illustrate this maturational process.6,7
In the United States, where demand governs a free-market and profit-driven system, the process of introducing a new medical technology to the marketplace is often chaotic and influenced by consumer demand, insurance and governmental payment systems, and the cost of product development. The enormous expenditure of industrial investment in research and development, preclinical and clinical trials, and marketing to both physicians and consumers begs to be recouped in patents guarding exclusivity and robust sales. Full description and analysis of this multifaceted, complex economic engine is beyond the scope of this chapter; however, all medical devices and technologies, regardless of the forces guiding them to market and their relative clinical merits, must clear the standards set by the US Food and Drug Administration (FDA).
Sponsors and investigators developing new technology for clinical purposes must focus their studies and trials on satisfying the requirements of the FDA. Initially, all products are subject to a premarket approval system, which evaluates their safety and effectiveness for a specific set of indications. Demonstrating this safety and effectiveness requires valid scientific evidence for the specific indications of use, which in most circumstances is obtained through human clinical studies. A clinical study must primarily take into account the indication for which clearance or approval is being sought. Specifically, the study must support that product’s safety and effectiveness for a given indication. If the data do not fully support the indication for which clearance or approval is being sought, the FDA will limit the indication for use to those indications for which valid scientific evidence exists.
Unlike the approval process of pharmacologic medications, which requires phase I, II, and III clinical trials,6,7 the approval process for new technologies or devices traverses a separate set of FDA regulations.8 In the 1960s, an explosion of technology introduced a large number of products to the market, some of which had the possibility of having risks for the patient. Various legislative measures were passed piecemeal by Congress until a more comprehensive solution was proposed by the Cooper Committee in 1970. This committee recommended a tiered regulatory system, in which devices posing a higher risk to patients would be subject to more demanding requirements. In 1976, the Medical Device Amendments, following the Cooper Committee’s recommendations, instituted a three-tiered risk-based system that is still in use today.
Class I devices are those not substantially important in preventing impairment of human health and that do not present a potentially unreasonable risk of patient injury. Examples of class I products include lead gonadal shields and x-ray grids. Class II devices present a greater risk of harm than class I devices and may be subject to additional regulation in the form of “special controls,“ which are applied to specific device types. Examples of class II products include higher technology products that do not by themselves maintain life, such as diagnostic devices, including CT, magnetic resonance, and ultrasound imaging units. Class III products include high-risk devices that are used to support or sustain human life. All products in this class are individually regulated and subject to a premarket approval process in which the manufacturer is required, like pharmacologic drugs, to establish the safety and effectiveness of the device before marketing it.8
If the FDA determines any new product to be “substantially equivalent“ to a pre-1976 product, the manufacturer can legally market the product under the terms of the previous product. Citations in peer-reviewed journals and expert opinions of physicians play an important role in the determination of substantial equivalence. Completely new products are automatically placed in the class III category and require clinical trials. This policy permits evolutionary changes, as witnessed in the advances seen in CT scanning, which the FDA permits to be marketed as a class II product because of its previous product and determination of substantial equivalence.8
Accuracy: Performance Characteristics of a Test
The simplest diagnostic test is one where the results of the study are used to classify patients into two groups according to the presence or absence of a disease. The discriminatory accuracy of a diagnostic test is commonly assessed by measuring how well it correctly identifies subjects who are known to be diseased and subjects who are known to be nondiseased.9,10 These data are commonly summarized in a 2 × 2 table of test results versus true disease state (Fig. 12–1). In the medical literature, measurement of the discriminatory accuracy of a test is expressed as the test’s sensitivity and specificity. Sensitivity is the proportion of true positives that are correctly identified by the test. Specificity is the proportion of true negatives that are correctly identified by the test. Expressed numerically, where TP = true positives, FP = false positives, FN = false negatives, and TN = true negatives, the sensitivity, or true-positive rate (TPR), is determined by the following equation: TP/TP + FN. The specificity, or true-negative rate (TNR), is determined by the following equation: TN/TN + FP. The false-positive rate (FPR), or the probability of a type I error, is determined by the following equation: 1 –specificity. Finally, the false-negative rate (FNR), or the probability of a type II error, is determined by the following equation: 1 –sensitivity.

A test that is highly sensitive will be positive in nearly all people with the disease state. Likewise, a negative result will make the chance of a subject having the disease unlikely. This is in contradistinction to a specific test. A highly specific test will be negative in virtually all patients without the condition, and a positive test makes the diagnosis highly likely.
A useful graphical summary of discriminatory accuracy is a plot of sensitivity versus FPR as the decision limit varies, which is called the receiver operating characteristic (ROC) curve (Fig. 12–2). The upper left corner of the graph represents perfect discrimination (TPR = 1 and FPR = 0), whereas the diagonal line where TPR equals FPR represents discrimination no better than random chance. The ROC curve is useful for comparing diagnostic tests, in particular, medical imaging techniques. The area under the ROC curve is a single number that summarizes what the ROC curve represents graphically.
FIGURE 12–2
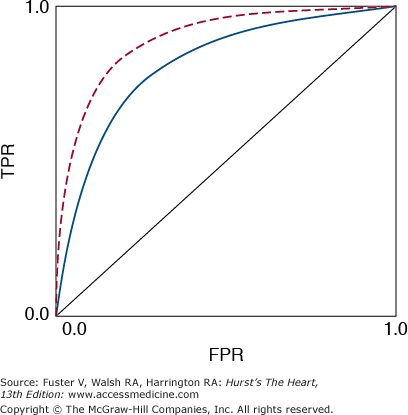
Receiver operating characteristic (ROC) curve. The red dashed line, with a larger area under the curve, represents a superior discriminatory test compared with the blue solid line. The dotted central diagonal line represents discrimination no better than random chance. TPR, true-positive rate; FPR, false-positive rate.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


