(1)
Department of Medicine, Cardiovascular Research Institute, University of Vermont College of Medicine, Colchester, VT, USA
Deceased
Abstract
Diabetes, insulin resistance, and heart disease are extraordinarily common. Furthermore, they are inextricably linked. The nature, cause-consequence connections, pathogenetic features, and diagnostic and therapeutic implications are the focus of this chapter. The seminal forms of heart disease, hypertensive heart disease, heart failure, and coronary artery disease, are those most ineluctably conflated with diabetes and insulin resistance. Numerous pathogenetic links are responsible. Though prevention and therapy of each are concordant with principles of management applicable to the same disorders in the general population, specific therapeutic considerations apply in patients with insulin resistance. Thus, knowledge of insulin resistance and diabetes is essential in informing treatment of heart disease. The panoply of therapeutics now available for treatment of diabetes provides powerful opportunities for amelioration of cardiovascular sequelae. Accordingly, the truly effective physician must function as both a diabetologist and a cardiologist to maximize the capacity to confer benefit to patients served. This chapter is designed to provide the information needed to facilitate achieving the necessary duality.
Keywords
DiabetesHeart failureHypertensionCoronary artery diseaseMetabolic syndromeInsulin resistanceAbbreviations
A1C
Hemoglobin A1c
ACE
Angiotensin-converting enzyme
ACS
Acute coronary syndrome
AGE
Advanced glycation end product
ARB
Angiotensin II receptor blocker
CABG
Coronary artery bypass grafting
CAD
Coronary artery disease
CAN
Cardiovascular autonomic neuropathy
CDE
Certified diabetes educator
CPT-1
Carnitine palmitoyltransferase-1
CRP
C-reactive protein
CVD
Cerebral vascular disease
DAG
Diacylglycerol
FA
Fatty acid(s)
FFA
Free fatty acid(s)
GLUT4
Glucose transporter 4
HDL
High density lipoprotein
HGO
Hepatic glucose output
IDL
Intermediate density lipoprotein
IP
Insulin-providing
IS
Insulin-sensitizing
LADA
Latent autoimmune diabetes in adults
LCAC
Long-chain acylcarnitine
LDL
Low density lipoprotein
LPL
Lipoprotein lipase
MI
Myocardial infarction
MODY
Maturity onset diabetes of the young
PAD
Peripheral arterial disease
PAI-1
Plasminogen activator inhibitor-1
PCI
Percutaneous coronary intervention
PKC
Protein kinase C
RAAS
Renin-angiotensin-aldosterone system
RAGE
Receptor for advanced glycation end products
TZD
Thiazolidinedione
UPA
Urokinase-type plasminogen activator
VLDL
Very low density lipoprotein
Introduction
Some Definitions
The sine qua non of the disorders called diabetes mellitus (diabetes) is physiologically inappropriate hyperglycemia. Elevation of concentrations of hemoglobin A1c (A1C) in blood (a time-integrated manifestation of hyperglycemia) is an additional diagnostic criterion. Classically, elevation of fasting blood glucose or impaired glucose tolerance (now called prediabetes to emphasize progression) have been the pivotal diagnostic criteria. The latter applies to prandial glycemia (2 h after eating) or to decreases in clearance of glucose administered intravenously. Episodic postprandial hyperglycemia per se (detectable by measuring serum 1,5-anhydroglucitol in view of its decrease with even transient hyperglycemia and consequent glycosuria) can be another criterion [1]. Current numerical values for diagnostic criteria are listed in Table 6.1 [2, 3]. Prediabetes is present when more modest changes obtain. It generally progresses to frank type 2 diabetes over many years. Approximately 19 million Americans have diabetes with another 7 million afflicted, but undiagnosed; 79 million (sic) have prediabetes [4].
Table 6.1
Diagnostic criteria for prediabetes and diabetes
Criterion | Prediabetes | Diabetes |
|---|---|---|
A1C | ≥6.5 % | |
Fasting plasma glucose (FPG) after minimum 8-h fast | 100–125 mg/dL (5.5–6.9 mmol/L) | ≥126 mg/dL (7.0 mmol/L) |
2-h plasma glucose during an oral glucose tolerance test | 140–199 mg/dL (7.7–10.9 mmol/L) | ≥200 mg/dL (11.1 mmol/L) |
Random plasma glucose with other symptoms of diabetes | ≥200 mg/dL (11.1 mmol/L) |
Two fundamental types of diabetes exist [5]. Type 1, previously often called juvenile or early onset diabetes, is attributable to deficiency of insulin. It reflects failure of pancreatic beta cells to elaborate insulin. Most type 1 diabetes is caused by autoimmune destruction of pancreatic beta cells although numerous other causes may pertain such as drug induced inflammatory pancreatitis. The sequelae of type 1 diabetes constitute what are called the microvascular complications of the disorder and include primarily retinopathy, neuropathy, and nephropathy. Their incidence and severity are linked directly to the magnitude and persistence of hyperglycemia. Uncontrolled, type 1 diabetes can cause ketoacidosis secondary to a flood from adipose tissue of free fatty acids (FFA) and their oxidation products. Because hyperglycemia associated with type 2 diabetes can give rise to the same microvascular complications that occur with type 1, it is imperative that stringent glycemic control be a therapeutic objective in all patients affected with diabetes, regardless of whether it is type 1 or type 2. Patients with type 1 diabetes can develop insulin resistance (sometimes called double diabetes), in part because insulin use can lead to obesity, which is in turn a cause of insulin resistance. In them, the metabolic derangements typical of type 2 diabetes will accompany the hallmark hyperglycemia [6].
Type 2 diabetes typically begins in adult life. It is attributable to resistance to the actions of insulin. As Reaven [7, 8], DeFronzo [9], and others have elucidated, insulin resistance is a tissue specific phenomenon. It gives rise to compensatory hyperinsulinemia, perhaps better called dysinsulinemia, that can spawn deleterious consequences. The insulinemic response is flawed in magnitude and timing. The first phase of postprandial elaboration of insulin in response to glucose is blunted or absent [10]. As long as the pancreas can compensate with sufficiently vigorous elaboration of insulin, frank diabetes does not ensue. Over time, however, the resistance to insulin in diverse tissues can no longer be obviated by compensatory dysinsulinemia because of beta cell failure caused in part by lipotoxicity, glycotoxicity, and genetic determinants. Frank diabetes then appears [11].
Numerous varieties of type 2 diabetes exist, and numerous molecular mechanisms can lead to insulin resistance. Maturity onset diabetes of the young (MODY) [12], is characterized by insulin resistance (type 2 diabetes) in youth. Conversely, type 1 diabetes can occur in adults (e.g., latent autoimmune diabetes in adults, LADA) [13]. Because type 2 diabetes constitutes more than 90 % of cases of diabetes in the population as a whole, it will be the focus of the discussion that follows unless noted otherwise.
Type 2 Diabetes
The cardiovascular sequelae of diabetes are linked closely not only to type 2 diabetes but also to insulin resistant states per se including the so-called metabolic syndrome. Targets for glycemic control, an imperative in type 2 as well as type 1 diabetes, are listed in Table 6.2. Because insulin resistance is intimately connected to obesity, pillars of management include dietary measures (caloric restriction and a “heart healthy” diet) and regular exercise. The help of a certified diabetes educator (CDE) is invaluable. Oral and injectable non-insulin hypoglycemic agents, incretins, and insulin, along with other measures titrated upward and in combination over time to achieve target concentrations of glucose and A1C are generally required [14].
Table 6.2
Targets for management of hyperglycemia
General glycemic targets | A1C <7.0 % |
Mean plasma glucose ~150 to 160 mg/dL (~8.3 to 8.9 mmol/L) | |
Preprandial plasma glucose <130 mg/dL (7.2 mmol/L) | |
Postprandial plasma glucose <180 mg/dL (10.0 mmol/L) | |
Individualized targets | More stringent control in younger, healthier patients (A1C target of 6.0–6.5 %) |
Less stringent control in older patients, those with comorbidities, and those prone to hypoglycemia or with hypoglycemia unawareness (A1C target of 7.5–8.0 %) | |
Treatment goal | The lowest A1C possible with avoidance of hypoglycemia |
Obesity and Diabetes
Insulin resistance precedes frank diabetes for years to decades. It is strongly associated with central obesity [15–19]. One biochemical factor of interest in the association, adiponectin, is elaborated from adipose tissue and known to enhance insulin signaling in muscle and liver. Its release is inverse to the mass of visceral fat [20]. Thus, its concentrations in blood decrease with central obesity and thereby contribute to insulin resistance. In addition, increases in visceral fat lead to release of inflammatory cytokines and diminished elaboration of leptin, both of which can impair insulin signaling.
Increased concentrations of circulating FFA associated with central obesity can inhibit the activity of the glucose transporter 4 (GLUT4) in skeletal muscle and hence glucose utilization. The increased FFA and glucose lead to accumulation of diacylglycerol (DAG) and triglycerides. FFA alone or in combination with DAG can activate protein kinase C (PKC) in muscle [21]. The result is serine phosphorylation (instead of the physiologic tyrosine phosphorylation) and hence inactivation of the insulin receptor, activation of the transcription factor NF-kB, generation of oxygen-centered free radicals by activation of NADPH oxidase, and consequent intensification of inflammation induced by pro-inflammatory cytokines.
In liver, insulin inhibits glycogenolysis [22] and gluconeogenesis, [23] leading to decreased hepatic glucose output (HGO). Consequently, insulin resistance increases HGO and contributes to hyperglycemia. Increased concentrations of FFA attenuate the effects of insulin that inhibit glycogenolysis and gluconeogenesis in liver and glucose transport in muscle thereby exacerbating metabolic manifestations of insulin resistance [24].
Insulin Resistance
Pivotal biological effects of insulin among a myriad of others are activation of acetyl-CoA carboxylase and lipoprotein lipase (LPL) (Fig. 6.1). The former converts acetyl-CoA to malonyl-CoA, the first step in fatty acid (FA) synthesis [25]. Through sequential addition of 2 carbon fragments, long-chain FA are synthesized and ultimately converted to very low density lipoprotein (VLDL) or stored as triglycerides in liver and adipose tissue [26]. Malonyl-CoA allosterically binds to the enzyme carnitine palmitoyltransferase-1 (CPT-1) and inhibits CPT-1 directly. Accumulation of long-chain acylcarnitine (LCAC) results because of the direct inhibition of CPT-1. In addition, the LCAC inhibits the enzyme CPT-1 secondarily. Accordingly, the transfer of long-chain FA into mitochondria is blocked. Thus, they do not undergo conversion to long-chain acylCoA and beta oxidation. Consequently, activation of acetyl-CoA carboxylase by insulin favors FA synthesis and diminishes FA oxidation, which are in turn dependent upon the mitochondrial shuttle system involving CPT-1 and -2.
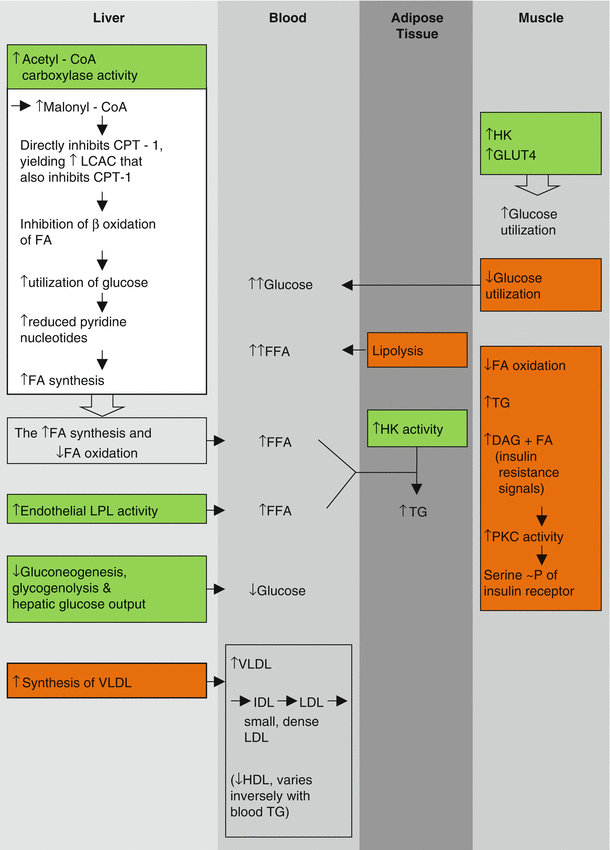
Fig. 6.1
Selected actions of insulin  and prominent changes with insulin resistance
and prominent changes with insulin resistance  . CPT-1 carnitine palmitoyltransferase-1, DAG diacylglycerol, FA fatty acid, FFA free fatty acid, GLUT4 glucose transporter 4, HK hexokinase activity, LCAC long-chain acylcarnitine, LPL lipoprotein lipase, ~P phosphorylation, PKC protein kinase C, TG triglyceride
. CPT-1 carnitine palmitoyltransferase-1, DAG diacylglycerol, FA fatty acid, FFA free fatty acid, GLUT4 glucose transporter 4, HK hexokinase activity, LCAC long-chain acylcarnitine, LPL lipoprotein lipase, ~P phosphorylation, PKC protein kinase C, TG triglyceride
and prominent changes with insulin resistance . CPT-1 carnitine palmitoyltransferase-1, DAG diacylglycerol, FA fatty acid, FFA free fatty acid, GLUT4 glucose transporter 4, HK hexokinase activity, LCAC long-chain acylcarnitine, LPL lipoprotein lipase, ~P phosphorylation, PKC protein kinase C, TG triglycerideWhen insulin resistance is present, the converse processes occur. FA and triglycerides are elaborated into blood; VLDL synthesis is increased in the liver; and the typical dyslipidemia ensues characterized by elevated concentrations of VLDL, triglycerides, and FFA in blood in association with reciprocally diminished concentrations of high density lipoprotein (HDL) [27, 28], a diminution known to be atherogenic.
Insulin potentiates uptake, phosphorylation, and oxidation of intracellular glucose in skeletal muscle. It suppresses prevailing concentrations of FFA and augments activity of hexokinase and the GLUT4 transporter facilitating utilization of glucose in muscle (oxidation of glucose and FFA are well known to be reciprocal). Conversely, insulin resistance leads to increased FA oxidation at the expense of utilization of glucose, a contributor to hyperglycemia.
Gerald Shulman’s group has provided intriguing information regarding the biochemical nature of insulin resistance [29, 30]. Insulin resistance is seen in skeletal muscle of insulin-resistant offspring of patients with type 2 diabetes. It is characterized by diminished utilization of glucose and decreased oxidation of FA. The fundamental defect appears to be a decrease in number or function of mitochondria. Increased intracellular FA and glucose lead to accumulation of DAG and triglycerides. DAG can activate PKC, known to yield serine phosphorylation as opposed to the physiological tyrosine phosphorylation, and hence inhibition of the insulin receptor. This may contribute to further decreases in glucose utilization, hyperglycemia, and the consequent initially compensatory hyperinsulinemia that precede development of diabetes for many years [31].
The action of insulin on LPL is tissue specific (Fig. 6.1). In adipocytes and capillary endothelium, insulin activates the enzyme. The result is shuttling of triglyceride-derived FFA to adipose tissue where it is stored as triglycerides diminishing lipolysis [27]. When insulin resistance is present, lipolysis is augmented and concentrations of FFA in plasma increase. In the liver, the increased FA augments synthesis of VLDL and increases both gluconeogenesis and glycogenolysis, normally physiologically inhibited by insulin [32–34]. Consequently, HGO increases, contributing to hyperglycemia. Thus, insulin resistance in vivo is characterized by elevated concentrations in blood of not only glucose and VLDL but also triglycerides and small, dense forms of low density lipoproteins (LDL) and intermediate density lipoproteins (IDL) derived from VLDL.
Insulin resistance can be the consequence of numerous metabolic, hormonal, and molecular causes (Fig. 6.2) including elevated FFA (seen with visceral adiposity), anomalous insulin receptor serine phosphorylation (seen with the polycystic ovarian syndrome) [35, 36], impaired mitochondrial FA oxidation, and impaired intracellular insulin signaling in multiple pathways and at multiple sites.
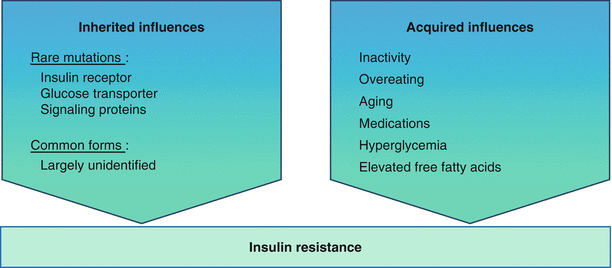
Fig. 6.2
Insulin resistance is caused by both inherited and acquired influences
The Metabolic Syndrome
Several clinically striking phenomena occur in conjunction with insulin resistance (Fig. 6.3). They include but are not limited to systemic arterial hypertension, central obesity, increased concentrations of triglycerides and VLDL, decreased concentrations of HDL, and impaired glucose tolerance. A procoagulant state, constrained fibrinolysis [37], impaired endothelial cell function, and microalbuminuria (all sometimes called non-traditional risk factors) are seen as well. Taken together, the constellation has been referred to variously as Syndrome X [7], the deadly quartet [38, 39], the deadly octet, or the metabolic syndrome. The constituent phenomena of the syndrome appear to aggregate more often than would be attributable to chance alone. It is not clear whether the aggregate cardiovascular risk exceeds that conferred by each combined, but the risks are at least additive [39–42]; the factors do identify patients at markedly increased risk of developing type 2 diabetes and cardiovascular disease (Fig. 6.4) [43]. Confusion has arisen in part because of semantics. Thus, cardiologists have used the term “Syndrome X” to refer to angina with angiographically normal coronary arteries. It is probably best applied to syndromes of insulin resistance [44–47].
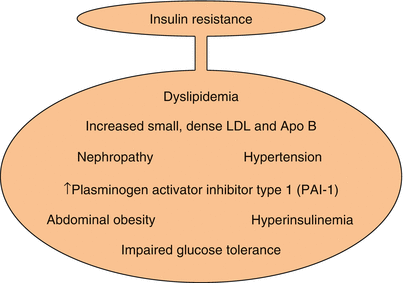
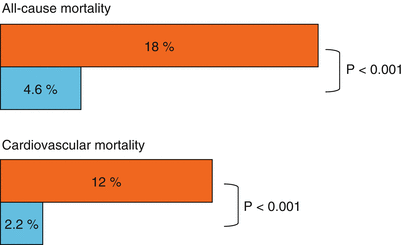
Fig. 6.3
Clinical phenomena seen in conjunction with insulin resistance. LDL low density lipoprotein
Fig. 6.4
Effect of the metabolic syndrome on cardiovascular and total mortality. Cardiovascular mortality was assessed for a median of 6.9 years in subjects with  and without
and without  the metabolic syndrome (Data used to construct the figure are from Ref. [42])
the metabolic syndrome (Data used to construct the figure are from Ref. [42])
and without the metabolic syndrome (Data used to construct the figure are from Ref. [42])The most commonly used clinical definitions of the metabolic syndrome include the presence of at least 3 of 5 abnormalities, including obesity, hypertriglyceridemia, low concentrations of HDL, systemic arterial hypertension, and insulin resistance with prediabetes or diabetes. The syndrome has been associated with a systemic inflammatory state reflected by increased C-reactive protein (CRP), non-alcoholic fatty liver disease, decreased adiponectin in blood [48], and altered platelet function [49]. The underlying insulin resistance and its sequelae certainly predispose to cardiovascular risk (Table 6.3). As Haffner has put it, the syndrome in association with type 2 diabetes confers a risk of myocardial infarction (MI) in those without previous MI equivalent to the risk of a subsequent MI in those without diabetes who have had a previous MI [50].
Table 6.3
Association of the metabolic syndrome with myocardial infarction
Odds ratio | 95 % CI | P value | |
|---|---|---|---|
Metabolic syndrome | 2.01 | 1.53–2.64 | <0.0001 |
Individual syndrome components | |||
Abdominal obesity | 1.15 | 0.86–1.54 | 0.3475 |
Hypertriglyceridemia | 1.51 | 1.04–2.20 | 0.0311 |
Low HDL concentration | 1.41 | 1.03–1.95 | 0.0353 |
Hypertension | 1.42 | 0.94–2.15 | 0.0947 |
Insulin resistance | 1.25 | 0.92–1.71 | 0.1461 |
Cardiovascular Manifestations of Type 2 Diabetes and the Metabolic Syndrome
Only relatively recently have diabetes and the metabolic syndrome been uniformly recognized by cardiologists to be risk factors for heart disease [51]. The data supporting the relationship are compelling [50, 52–59]. The metabolic syndrome, even in the absence of frank diabetes, is a risk factor also for heart failure with or without associated MI [60].
Diabetes has been implicated, at least anecdotally, in accelerating or causing numerous kinds of cardiovascular disease, including valvular heart disease, coronary arterial calcification, and cardiomyopathies. However, it is unequivocally linked to 3 major types of heart disease—hypertensive cardiovascular disease, heart failure, and coronary artery disease (CAD). More than 80 % of people with diabetes die as a result of cardiovascular disease [61].
Hypertension
Infusion of insulin induces endothelin-1 dependent hypertension in animals [62–64]. The diabetic state is manifested in part by oxidative stress attributable to excessive generation of oxygen-centered free radicals from oxidation of glucose and activation of NADPH dehydrogenase. These in turn impair endothelial cell function and endothelial cell mediated vasodilation and intensify the systemic inflammatory state. Reaven and colleagues [65] showed in patients with idiopathic systemic arterial hypertension (previously referred to as essential hypertension based on the false premise that the hypertension was “needed” to propel an adequate supply of blood through obstructed vasculature) that high concentrations of insulin in plasma were prevalent. Insulin resistance and its associated dysinsulinemia are associated with increased activity of the renin angiotension aldosterone system (RAAS) and the central-sympathetic nervous system; vasoactive syndromes mediated by endothelin and inhibited by nitric oxide [47]; and augmented salt sensitivity and hypervolemia [66]. Accordingly, obese subjects with insulin resistance manifest a particularly high incidence of systemic arterial hypertension [67].
Management of hypertension in patients with diabetes does not differ substantively from management of hypertension in the population as a whole. However, it must be particularly aggressive because hypertension can accelerate CAD and heart failure, both of which are prominent with diabetes.
Heart Failure and Diabetes
Heart failure associated with diabetes is typically more frequent in females, associated with preserved systolic function, and associated with hypertension [68]. It may be independent of an ischemic cardiomyopathy. Its incidence is more than double that in those without diabetes regardless of age (Fig. 6.5) [69, 70].
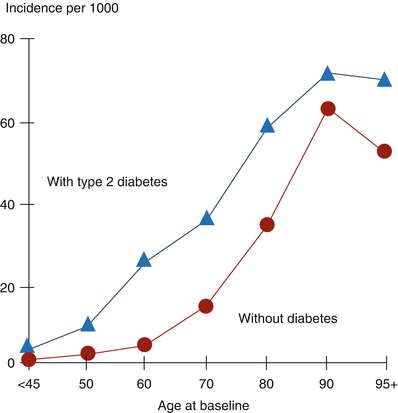
Fig. 6.5
Incidence of heart failure (HF) is generally more than doubled in patients with type 2 diabetes. In this study, medical records of subjects with type 2 diabetes but without HF at baseline (n = 8,460) were compared with records for age- and sex-matched subjects without diabetes and without HF (n = 9,156). Over a 30-month period, 650 subjects with diabetes (7.7 %) developed HF compared with 314 of the subjects without diabetes (3.4 %) (Data used to construct the figure are from Ref. [68])
In patients who sustain an acute coronary syndrome (ACS), diabetes confers a markedly increased risk for development of heart failure subsequent to the index event [53]. Furthermore, it accounts for a marked increase in overall mortality over 24 months. Paradoxically, diabetes, rather than the angiographically detectable extent of CAD, is the most powerful predictor of death and MI in patients treated initially for an ACS [71, 72]. Ironically, heart failure per se can induce insulin resistance likely to be attributable in part to counter-regulatory hormones and cytokines [73].
Acoustic properties of myocardium are abnormal in patients with diabetes despite preservation of left ventricular ejection fraction [74], abnormalities that have been referred to as subclinical cardiomyopathy [75]. In patients who sustain an acute MI the incidence of heart failure is greater when diabetes is present when results are normalized for infarct size [76]. In experimental animals subjected to MI, subsequent fibrosis is more extensive when diabetes is present [77]. The increase is caused in part by increased plasminogen activator inhibitor-1 (PAI-1), secondary to insulin resistance. PAI-1 is known to be profibrotic in the heart [78]. The altered myocardial backscatter in patients with type 2 diabetes may reflect increased fibrosis secondary to PAI-1 and direct effects of augmented activity of the RAAS [79–81]. Results in the EPHESUS trial substantiated the value of inhibition of this system [82] in a large population [83].
Diabetes can cause a cardiovascular autonomic neuropathy (CAN) [84]. CAN may increase the risk of sudden cardiac death associated with stringent glycemic control because of the absence of adequate counter-regulatory responses to hyperglycemia supporting cardiovascular function [85–88]. The absence of reduction of morbidity and mortality with intensive anti-glycemic therapy in clinical trials, such as ADVANCE, VADT, and ACCORD, may reflect, in part, adverse consequences of CAN.
Management of Patients with Heart Failure and Diabetes
Management is generally the same as that for heart failure in patients without diabetes. Thus, amelioration of identifiable known causes (such as ischemic cardiomyopathy) with appropriate interventions, reduction of left ventricular afterload, treatment of hypertension, and the use of diuretics are mainstays of therapy. Some specific considerations merit attention, however.
Historically, heart failure was managed by administering agents with positive inotropic effects, such as digitalis glycosides, to patients with impaired ventricular performance. Ultimately, the failure of this approach to improve long-term prognosis led to a profound shift in focus. Before the advent of diuretics, fluid overload was sometimes managed by desperate use of invasive lymphatic drainage with Southey tubes [89]. The first diuretics were injectable mercurial agents that were associated unfortunately with adverse effects on the kidney. Thiazide diuretics were discovered serendipitously after the advent of sulfonamides for the treatment of infection. Sulfonamide-associated diuretic effects led to molecular tailoring of similar agents without antibiotic properties but with marked diuretic effects. Reduction of left ventricular afterload with the use of blockers of the RAAS was initially recognized to be beneficial in heart failure associated with acute mitral regurgitation [90], and developed extensively by Jay Cohn’s group in the treatment of heart failure of diverse etiologies [91, 92]. It soon became an established pillar of therapy [93–96].
Historically, it was felt that agents that reduced contractility, such as beta adrenergic blocking agents, were contraindicated in patients with heart failure. However, following the pioneering work of Hjalmarson’s group [97, 98] and numerous clinical trials (reviewed in [99]), therapy with beta blockers became a standard of care in patients with heart failure whether or not they had diabetes. It had been feared that beta blockers would cause hypoglycemia in patients with diabetes, but in general, they have been well tolerated [100]. An exception may be in those patients with bradycardia or CAN in whom the risk of hypoglycemia may be particularly high.
A number of agents may ameliorate pathophysiologic factors contributing to heart failure in patients with diabetes, including:
diuretics;
calcium channel blockers;
beta adrenergic blocking agents;
afterload reducing agents including angiotension-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs);
anti-arrhythmic agents;
agents with positive inotropic effects under specific circumstances such as imminent pump failure; and
anticoagulants and antiplatelet agents.
Agents that inhibit the deposition or activity of advanced glycation end products (AGEs) and their receptor (RAGE) [101]; antiglycemic agents that exert favorable effects on ventricular performance including incretins (glucagon-like peptide 1 (GLP-1) agonists and dipeptidyl peptidase-4 (DPP-4) antagonists); and insulin sensitizers to attenuate potentially deleterious overexpression of PAI-1 with its propensity to impair fibrinolysis and induce cardiac fibrosis [78] may be beneficial. Preclusion of hyperosmotic effects of hyperglycemia, diminution of autophagy [102], enhancement of myocardial energetics with agents such as pyruvate [103, 104], and amelioration of hypertension with antagonists of endothelin [62–64] are being explored as potential therapeutic approaches as well.
Coronary Artery Disease
Viewed most simplistically, CAD, like peripheral arterial disease (PAD) and cerebral vascular disease (CVD), is a manifestation of atherosclerosis. Several mechanisms may explain how diabetes accelerates atherosclerosis or can modify its manifestations. The dyslipidemia typical of insulin- resistant states and type 2 diabetes undoubtedly plays a role. Hypertension can accelerate atherogenesis, and is, of course, a key concomitant of insulin resistance. The association between obesity and atherosclerosis is well recognized.
Somewhat counterintuitively, it is not entirely clear whether hyperglycemia per se is a major, direct contributor to atherosclerosis in association with diabetes [105]. Patients with type 1 diabetes do not necessarily manifest accelerated macrovascular disease in the absence of dyslipidemia. Stringent glycemic control does, of course, normalize insulin resistance-associated determinants of CAD thereby decreasing risk [106]. Stringent suppression of hyperglycemia in studies such as the UKPDS did lead to modestly favorable effects on the incidence of events associated with macrovascular disease as well as markedly favorable effects on the progression of microvascular disease (Fig. 6.6) [107].
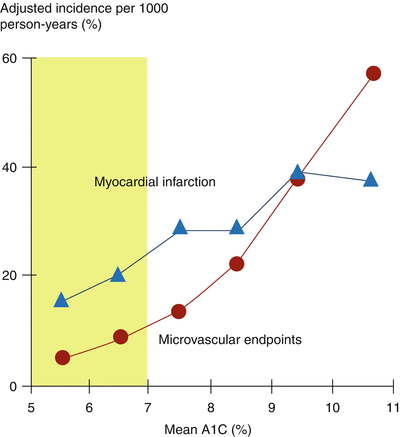
Fig. 6.6
Effect of A1C reduction to <7.0 % on incidence of macrovascular disease and progression of microvascular disease. Mean data (point estimates) from the UKPDS, N = 4,585 (Data used to construct figure are from Ref. [105])
In considering atherosclerotic CAD, it is important to differentiate between biologically stable and unstable plaques. Plaques that are richly endowed with vascular smooth muscle (thought to migrate from the tunica media to the neointima during the evolution of atherosclerosis) are biologically stable and underlie slowly evolving CAD. A clinical hallmark of such plaques is clinically stable angina pectoris. Atherosclerotic plaques associated with sudden cardiac death and the precipitation of ACS in general differ markedly from those associated with stable CAD. They are biologically unstable, lipid laden, relatively devoid of vascular smooth muscle cells, covered with thin as opposed to thick fibrous caps, friable [108], and vulnerable to rupture thereby initiating catastrophic intravascular thrombosis and complete coronary occlusion precipitating ACS, including sudden cardiac death (Fig. 6.7).
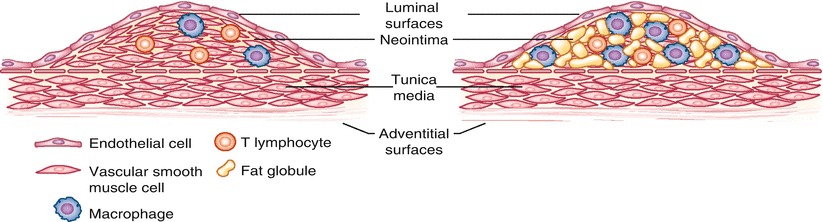
Fig. 6.7
Depiction of biologically stable (left) and unstable (right) plaques
Patients with type 2 diabetes have angiographically more extensive macrovascular coronary disease compared with that seen in those without diabetes [109–111]. Distal disease and small vessel CAD are more prevalent [109]. The morphology of plaques differs from that in patients without diabetes and is typical of vulnerable plaques. One factor that may be responsible is augmented mural expression of PAI-1 [108, 112, 113]. It is well known that migration of diverse types of cells is facilitated by cell surface expression of activators of plasminogens (particularly urokinase-type plasminogen activator (UPA)). It is well established also that insulin augments expression of PAI-1 within vessel walls and that PAI-1 can inhibit cell migration. Accordingly, it is not surprising that augmented expression of PAI-1 typical of type 2 diabetes can inhibit migration of vascular smooth muscle cells from the tunica media to the neointima thereby predisposing to the evolution of plaques relatively devoid of them and manifesting features of vulnerability to rupture. In concert with a constrained fibrinolytic system as well as a prothrombotic state, the anomalous evolution of atherosclerotic plaques appears to account in part for the markedly increased risk of ACS seen with diabetes [114].
Despite the advent sequentially of coronary care units, the use of thrombolytic agents, the widespread utilization of primary percutaneous coronary intervention (PCI), and advances in coronary artery bypass grafting (CABG), patients with diabetes have continued to exhibit an approximately two-fold greater incidence of ACS and mortality (Fig. 6.8) [61], restenosis following PCI, and virtually all other manifestations of CAD [53, 115–125] compared with corresponding incidences of the same phenomena in patients without diabetes. The same appears to be true for patients with the metabolic syndrome in the absence of frank diabetes [126–128].
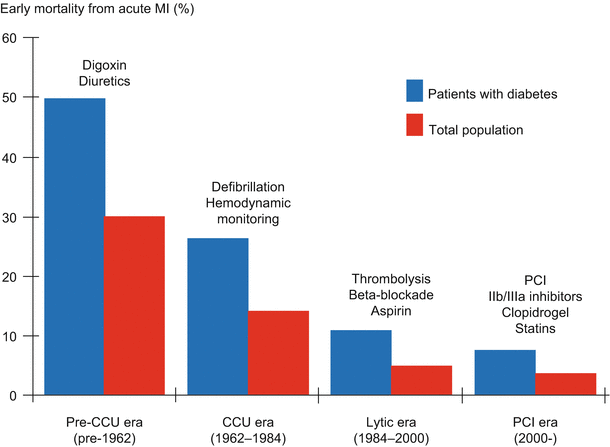
Fig. 6.8
Diabetes doubles risk of mortality with myocardial infarction (MI) despite advances in cardiac care (Data used to construct the figure are from Richard Nesto by personal communication and adapted from Ref. [59])
Diabetes, Thrombosis, and Fibrinolysis
Sequelae of macrovascular disease are the major contributors to mortality in patients with diabetes [129–131] and are often precipitated by thrombosis [132]. Several mechanisms may link thrombosis to insulin resistance, including augmented numbers and activity of platelets; elevated concentrations in blood of procoagulant proteins; decreased concentrations in blood of antithrombotic factors such as anti-thrombin III activity (also called heparin co-factor activity); and decreased concentrations in blood of protein C (reviewed in [133]). Evolution of micro- to macro-thrombi is augmented by a relative imbalance between thrombosis and fibrinolysis favoring thrombosis. Platelet reactivity is increased in patients with diabetes secondary to hyperosmotic effects of hyperglycemia on platelet function and altered platelet surface receptor function attributable to increased glycation. The increased platelet reactivity predisposes to an increased risk of ACS. Accordingly, patients with diabetes and insulin resistance are prone to exhibit abnormalities not only of the fibrinolytic system mediated by increased PAI-1 [134–145] but also of platelet activation [146] and procoagulation. Taken together, these phenomena appear to contribute to the increased incidence of ACS seen with diabetes [112, 113].
Fibrinolysis is constrained in vitro by insulin, pro-insulin, and pro-insulin split products as well as by metabolic factors associated with insulin resistance in vivo. Insulin inhibits the rate of degradation of PAI-1 messenger RNA [147] and hence augments expression of PAI-1. Both FFA and VLDL increase expression of PAI-1 [143]. Hyperglycemia per se may inhibit fibrinolysis by increasing expression of PAI-1 in endothelial cells [138].
Increased concentrations of PAI-1 in blood have been observed consistently in patients with type 2 diabetes [148–150] and in those with the insulin resistance seen with the polycystic ovarian syndrome [35, 36]. Administration of insulin, liposyn A, and glucose to normal human subjects augments concentrations of PAI-1 in blood [151]. Coronary atherectomy specimens from patients with diabetes exhibit increased expression of PAI-1 [144].
In part because of these observations, the BARI 2D trial was undertaken to determine whether an insulin-sensitizing (IS) as opposed to an insulin-providing (IP) pharmacologic strategy would lead to differences in the expression of biomarkers indicative of activity of the fibrinolytic system [152]. The IS treatment strategy was associated with a 5-year consistent diminution of the concentrations of PAI-1 antigen in blood and of PAI-1 activity. Thus, the results indicated that the balance between thrombosis and fibrinolysis had been shifted favorably toward fibrinolysis by the IS strategy (Fig. 6.9), portending favorable clinical benefit.
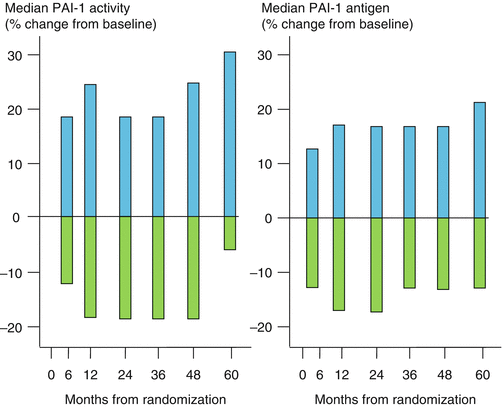
Fig. 6.9
Comparison of insulin-sensitizing  with insulin-providing
with insulin-providing  treatment strategies on PAI-1 activity and PAI-1 antigen over 5 years in the BARI 2D trial. The treatment groups had the same baseline values for PAI-1 activity (16 AU/ml) and antigen (23 ng/ml) (Data used to construct the figure are from Ref. [150])
treatment strategies on PAI-1 activity and PAI-1 antigen over 5 years in the BARI 2D trial. The treatment groups had the same baseline values for PAI-1 activity (16 AU/ml) and antigen (23 ng/ml) (Data used to construct the figure are from Ref. [150])
with insulin-providing treatment strategies on PAI-1 activity and PAI-1 antigen over 5 years in the BARI 2D trial. The treatment groups had the same baseline values for PAI-1 activity (16 AU/ml) and antigen (23 ng/ml) (Data used to construct the figure are from Ref. [150])Diabetes and the Vessel Wall
The BARI 1 trial [153] compared results obtained with CABG as opposed to PCI in patients with clinically unstable CAD requiring coronary intervention. In the trial as a whole, the outcomes following each intervention were comparable over 5 years of follow up. However, post hoc analysis showed that after CABG in the patients with diabetes, mortality was two-fold greater over 5 years. Furthermore, mortality following initial PCI was four-fold greater. The increased mortality after PCI was associated with late vasculopathy in the segments that had been subjected to the coronary intervention. This led us and others to hypothesize that the iatrogenic trauma of PCI in patients with diabetes led to adverse effects because of the susceptibility of their vasculature to develop plaques vulnerable to rupture and their predisposition to thrombosis [154]. The BARI 1 results led to the issuance of an NHLBI clinical alert urging caution in the use of coronary interventions in patients with diabetes. In fact, as discussed below, CABG may often be a favorable alternative to PCI in patients with diabetes, especially in those with complex multi-vessel disease.
Additional insights were provided by results from the BARI 2D trial [155] in patients with clinically stable CAD (as opposed to unstable CAD in the BARI 1 trial). Patients underwent a 2 × 2 factorial design randomization procedure. They were randomized to either optimal medical management alone or optimal management coupled with coronary intervention (CABG or PCI predicated on the preferences of the investigators evaluating the enrollment angiograms). They were randomized also to treatment with either an IP or IS regimen. Statistically significant differences in clinical outcomes (defined as all-cause mortality, MI, and stroke) were not apparent with respect to either randomization. However, hazard ratios were lower in patients treated with the IS strategy, particularly in those who were randomized also to early intervention and who underwent CABG. These results were consistent with striking and highly statistically significant differences in biomarker profiles of patients treated with the IS compared with the IP strategy [152]. The IS strategy was associated with markedly diminished concentrations of PAI-1 in blood over the 5-year follow up interval (Fig. 9) accompanied by evidence of diminished thrombosis reflected by concentrations of fibrinopeptide A and a diminished intensity of the systemic inflammatory state reflected by high-sensitivity assays of CRP.
Treatment of CAD in Patients with Diabetes
Management of CAD in patients with diabetes is generally concordant with management of patients with CAD in the population as a whole. Reduction of risk has been amply and definitively demonstrated in numerous studies focusing on lifestyle modifications (especially diet and exercise and cessation of smoking), amelioration of dyslipidemia, control of hypertension, and control of hyperglycemia. Of particular interest is the striking benefit of control of blood pressure as seen in the seminal UKPDS study. Its impact was surprisingly even greater than that of glycemic control per se (Fig. 6.10) [156].
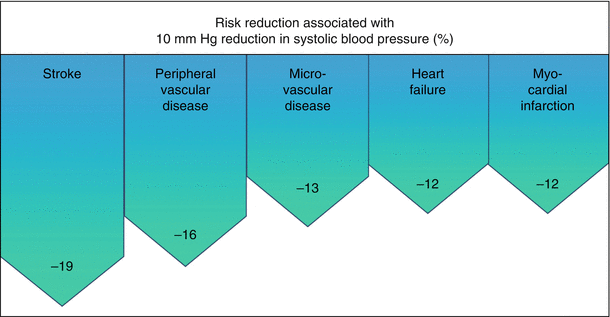
Fig. 6.10
Effect of control of blood pressure (BP) on diabetes-related complications (Data are from 3,642 patients followed as part of the UKPDS Ref. [154])
Diabetes is known to compromise clinical outcomes following successful PCI or CABG [122, 157–161]. Patients with diabetes have a higher incidence of “no (or low) reflow” following PCI for treatment of ST-segment MI [162]. In addition, they exhibit a greater incidence of the need for revascularization following initially successful PCI. In patients with diabetes and complex multi-vessel disease, CABG may be the preferred mode of intervention. Mortality following CABG compared with that after PCI has often been comparable in rigorously controlled studies, but the need for multiple revascularization procedures following PCI may justify CABG as the preferred intervention (reviewed in [163]) [160, 164–174]. The possibility that the relative benefits of drug eluting stents will soften this justification is being explored [175].
Diabetes compromises the benefits of coronary thrombolysis induced with fibrinolytic agents. It is associated with accelerated restenosis after coronary intervention, perhaps related at least in part to inhibition of apoptosis by the augmented intramural expression of PAI-1 [176]. Despite advances in the development of drug-eluting stents and other technologies, diabetes continues to confer an adverse prognosis after coronary intervention compared with that seen in patients without diabetes [163].
An issue that has been a contentious focus of recent study is the question of whether patients with diabetes and an ACS should be subjected in the intensive care setting to antiglycemic treatment designed to achieve stringent glycemic control. Benefit has been observed in some studies, including the DIGAMI trial [177], in critically ill patients in diverse settings [178, 179] but not in others by some of the same investigators [180], a difference possibly attributable to methodologic difficulties. The adverse effects of hypoglycemia may, in fact, militate against the wisdom of this approach [181, 182]. Thus, instead of normalization of glycemia to 80–110 mg/dL as in DIGAMI, a target of 140–180 mg/dL may be better [181–183].
Therapeutic Implications
Treatment of Hypertension
Management of hypertension in patients with diabetes should be aggressive (generally targeting blood pressure of 130/80 mm Hg or less). It should focus also on specific abnormalities likely to be present in patients with diabetes including hyperactivity of the RAAS. Accordingly, the use of ACE inhibitors, ARBs, and antagonists of aldosterone, such as eplerenone, should receive high consideration. Although beta blockers can induce hypoglycemia, particularly non-selective or beta-1 selective agents, the use of non-beta-1 selective agents in patients with diabetes is generally safe and effective [184, 185]. Diuretics are often indicated as well and generally well tolerated despite the possibility that thiazide diuretics can exacerbate hyperglycemia even though the phenomenon has less long-term adverse cardiovascular effects than does native diabetes [186]. Nevertheless, substitution of chlorthalidone for a thiazide may be advantageous. The aldosterone receptor antagonist triamterene has been employed, but it is not used widely. Agents under development such as antagonists of endothelin may ultimately prove to be useful.
Heart Failure
Patients with heart failure and diabetes are managed similarly to those with heart failure without diabetes. However, some specific considerations may apply. CAN, activation of the RAAS, and cardiac fibrosis as well as hypertension may be prominent features contributing to heart failure in patients with diabetes. Accordingly, the use of ACE inhibitors, ARBs, aldosterone antagonists, antagonists of the aldosterone receptor, rigorous control of hypertension, and the use of beta blockers merit particular consideration. Special caution is required in the use of beta blockers if CAN is present. Future studies may justify the use of novel agents such as those that degrade cross-linking induced by AGEs, inhibit activation of RAGE, or antagonize endothelin. The use of natriuretic peptides in patients with heart failure and profound pump dysfunction has been considered but not widely adopted.
Following the widely publicized and intensely criticized meta-analysis condemning rosiglitazone as an agent with increased risk of cardiovascular mortality in patients with diabetes [187], much clarification has emerged [152, 188, 189]. Furthermore, the original claims have been disavowed (“updated”) by the original authors [190]. Thiazolidinediones (TZDs) such as rosiglitazone activate sodium retention by the kidney [191] and can therefore exacerbate heart failure [192, 193]. However, a panoply of data demonstrate the safety of TZDs in patients in whom heart failure is not present [152, 194, 195]. Thus, the VADT, ACCORD, ADVANCE, RECORD, and BARI 2D trials showed no increased risk with rosiglitazone. Though early studies with troglitazone in experimental animals raised a specter of possible adverse effects on left ventricular function, studies in human subjects have consistently indicated no such adverse effects [192]. In fact, rigorously controlled prospective studies and trials have consistently supported the benefits of TZDs in patients with diabetes without heart failure and have failed to demonstrate adverse effects on cardiovascular mortality or all-cause mortality (Table 6.4). By contrast, favorable cardiovascular effects have been delineated (Fig. 6.11).
Table 6.4
Reductions in rates of adverse events during and within the 3 months (legacy) after completion of the BARI 2D trial demonstrate benefit of treatment of type 2 diabetes with thiazolidinediones
Outcome | Rate per 100 patient-years | RR | p value | |
|---|---|---|---|---|
Rosiglitazone | No thiazolidinedione | |||
Adverse events during treatment | ||||
Death | 1.88 | 2.56 | 0.77 | 0.08 |
Myocardial infarction (MI)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||