Development of Myocardial Structure and Function
Pamela A. Lucchesi
Aaron J. Trask
Rachel C. Childers
Richard L. Goodwin
Introduction
The structure and function of the myocardium undergoes dramatic changes during fetal life and in postnatal maturation to adulthood. The postnatal period is marked by extensive physiologic and metabolic remodeling with dynamic changes as the fetal heart adapts to birth and converts to adult function (1). These processes are regulated by a number of hormones, neurotransmitters, growth factors, and mechanical forces. At the cellular level, changes in gene transcription and alternative splicing (1), epigenetic modulation of DNA methylation (2), and microRNA (miR)-induced posttranscriptional gene expression programs (3,4) have emerged as key regulators of cardiac myocyte growth and phenotype. The coronary circulation is tightly coordinated with myocardial growth to ensure an adequate supply of oxygen and metabolic substrates. A complete understanding of the physiologic processes that regulate myocardial structure and function is a necessary prerequisite to understand the pathogenesis of congenital and acquired heart disease. While Chapter 1 in this volume provides a comprehensive discussion of the molecular and genetic determinants of heart development, this chapter describes the developmental and postnatal changes in cardiac structure, metabolic regulation, excitation–contraction (E-C) coupling, and growth/regeneration. The role of cell–cell and cell–matrix interactions is discussed. Postnatal changes in hemodynamic load, autonomic innervation, and hormonal status are summarized. The effects of these changes on myocardial systolic and diastolic dysfunction are also discussed.
The majority of studies on developmental changes in myocardial structure and function has been performed in zebrafish, chick embryos, and rodents, with some additional data taken from higher mammals and humans. While the process of E-C coupling is very similar, there is significant spatiotemporal variability in structural development among the different model species. Unless otherwise noted, the majority of the developmental changes described in this chapter will focus on data from rodent models and humans. Secondly, most of our understanding of developmental changes in myocardial structure and function is limited to the left ventricle (LV), and little is known about right ventricular (RV) development or physiologic interactions between the left and RV that occur during postnatal maturation (3).
Myocardial Structure
The heart begins functioning as a simple tube composed of only cardiac myocytes and endocardial cells. However, it quickly becomes a complex organ comprising multiple cell types that can be grouped into conducting, supporting, and functional cells (Fig. 4.1). The cellular constituents of the heart include cardiac myocytes, cardiac fibroblasts, endothelial cells, and vascular smooth muscle cells. The sinoatrial nodes are specialized myocytes responsible for action potential generation. The conducting cells, also derived from cardiac myocytes are mainly Purkinje fibers. Purkinje fibers propagate the action potential from the atrium to the ventricle. While cardiac myocytes are responsible for the mechanical function of the heart, they comprise only ∼30% of the total number of cells. Cardiac fibroblasts predominate in conferring structural integrity to the heart (5). Dynamic crosstalk between cardiac myocytes, cardiac fibroblasts, and the extracellular matrix (ECM) plays a crucial role in myocardial development and structural remodeling.
Cardiac Fibroblasts and the Extracellular Matrix
The cardiac fibroblast is the most abundant cell type present within the postnatal mature heart. They are the main cell type responsible for the ECM deposition and also dynamically remodel the ECM through the secretion of matrix metalloproteinases and their tissue inhibitors. Cardiac fibroblasts are derived from different cell lineages at different developmental stages. During embryonic development they are mesenchymal in origin and are derived from the proepicardium (PE). Fibroblasts also arise from the differentiation of bone marrow–derived circulating fibrocytes (6). In the neonatal and adult heart, cardiac fibroblasts arise from resident cells via epithelial–mesenchymal transformation and from bone marrow–derived cells (7). Thus, cardiac fibroblasts from the neonatal period are distinct from those in the adult myocardium and are also different from the ones that populate the heart following acute injury or chronic hemodynamic overload (8).
Cardiac fibroblasts play an extensive role in cardiac development and remodeling. During development, fibroblasts secrete a number of growth factors that promote cardiac myocyte proliferation. Matrix deposition during this period establishes a functionally competent ventricle, which provides structural stability necessary for transitioning from fetal to postnatal life (9). Following birth, cardiac fibroblasts double in the early postnatal mouse heart and increase ECM deposition and organization to provide tensile strength to the myocardial wall.
The ECM provides three-dimensional structural support to the heart, facilitates mechanical and chemical signaling during myocardial development, and helps to maintain normal homeostasis in the adult heart in response to physiologic stressors and injury. Components of the ECM include interstitial collagens, elastin, fibronectin, proteoglycans, glycoproteins, cytokines, growth factors, and proteases. The most abundant extracellular collagens are the fibrillar collagens type I and III, though a number of nonfibrillar collagens are also found in the ECM of the developing and adult myocardium. Elastic fibers are present in close association to collagen and are responsible for maintaining normal elasticity of the cellular framework. Fibronectin influences cellular properties, including cellular growth. Proteoglycans and glycoproteins appear to play a role in signaling and ECM turnover, and serve as a reservoir for latent growth
factors (e.g., transforming growth factor β [TGF-β], epidermal growth factor).
factors (e.g., transforming growth factor β [TGF-β], epidermal growth factor).
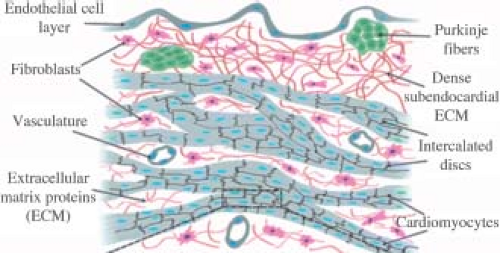 Figure 4.1 Schematic representation of the cellular and structural components of the ventricular myocardium, including myocytes, fibroblasts, blood vessels, and Purkinje fibers. The endothelial cell layers line the surface of the endocardium (top) and is supported by a layer of dense extracellular matrix (collagens, elastins, fibronectin, proteoglycans) secreted by interstitial cardiac fibroblasts (pink). The Purkinje fibers (green) are specialized cardiac muscle fibers located within this matrix that are responsible for electrical impulse propagation from the atrioventricular node to the ventricular myocardium. Cardiac myocytes are organized in myofibers and are electromechanically coupled by intercalated discs. Small blood vessels and capillaries are located adjacent to myofibers to provide nutrients, deliver oxygenated blood, and remove metabolic by-products. (Reproduced from Simmons CS, Petzold BC, Pruitt BL. Microsystems for biomimetic stimulation of cardiac cells. Lab Chip 2012;3235–3248.) (185) with permission from the Centre National de la Recherche Scientifique (CNRS) and The Royal Society of Chemistry.) |
The collagen network of the myocardium begins forming during fetal development (7). Cardiac fibroblasts become enmeshed in this network, which allows them to contract the endomysial collagen, exerting mechanical force on the myocytes. In addition, this organization allows fibroblasts to maintain structural integrity of the heart through cell–cell and cell–ECM interactions, as well as through proliferation and ECM degradation. In the adult myocardium, this network includes the epimysium that surrounds large groups of muscle fibers, the perimysium arising from the epimysium that surrounds smaller groups of muscle fibers and the endomysium, which tethers individual fibers to each other and the adjacent vasculature (Fig. 4.2) (10,11). In addition to acting as scaffolding for cells and vessels, the collagen network also coordinates the transmission of force generated by myocytes, serving as a viscoelastic medium facilitating compression and recoil properties of the tissue (12).
Cardiac fibroblasts are regulated by mechanical and molecular signals during cardiac development. Mechanical stimulation of fibroblasts causes a marked increase in integrin receptors and secretion of ECM proteins, cytokines, and growth factors. Angiotensin, endothelin, and TGF-β also stimulate matrix deposition, while the inflammatory cytokines interleukin-1β and tumor necrosis factor-α inhibit matrix deposition and promote matrix degradation by matrix metalloproteinases (13).
Basement Membrane
A specialized area of the matrix termed the basement membrane or basal lamina surrounds all cells in the myocardium except cardiac fibroblasts. An intact basement membrane is necessary for normal cardiac growth and maturation and plays an important role in postnatal cardiac myocyte sarcomerogenesis via activation of integrin-mediated signaling (14). The primary components of the basal lamina are laminin, type IV collagen, and the proteoglycan perlecan. Type IV collagen assembles into a sheet-like mesh and provides tensile strength. Type IV collagen is coated by perlecan, which binds to integrins and dystroglycans on the plasma membrane to stabilize the basement membrane structure to withstand mechanical forces. The basement membrane is the first ECM produced during embryonic development. Laminins are detected in the human heart as early as 8 weeks of gestation while collagen IV is accumulated postnatally (14,15). Perlecan is expressed at high levels throughout embryogenesis in the heart and required to ensure mechanical stability until cell–cell contacts have formed and matured (16).
Cardiac Myocytes
Cardiac myocytes are derived from two waves of anterior splanchnic mesoderm known as the primary heart field, which forms the primary heart tube. The second heart field makes a significant contribution of myocytes of the RV and the myocardium associated with the arterial pole of the heart (17). Cardiac myocytes have two major mechanistic functions: Force generation by myofibrils in response to E-C coupling and force transmission across cell bundles mediated by the integration of electromechanical signals at the intercalated disc. Myocytes also communicate with the ECM and other cardiac cells through costameres (see below).
Plasma Membrane
The plasma membrane (or sarcolemma) is the region of the cell that contains ion pumps, channels, and exchangers that contribute to action potential propagation, as well as maintenance of proper ionic and chemical gradients. The flow of ions controlled by these proteins is essential for proper myocyte function and directly regulates cellular contraction and relaxation. Numerous G-protein–coupled receptors, cytokine receptors, and growth factor receptors are located on the plasma membrane and are responsible for transducing changes in the local neurohormonal milieu into intracellular signals that regulate cell growth and function.
Na+ and Ca2+ ATPases and Exchangers
The Na+–K+ ATPase maintains the ionic gradient across the sarcolemmal membrane by pumping Na+ out of the cell and K+ into the cell against their respective concentration gradients. Since under physiologic conditions, three Na+ are removed from the cell but only two K+ ions are taken up per pump cycle, the ATPase generates a small outward current. Both the maintenance of the ionic gradient and the electrogenicity of Na+–K+ ATPase are required to maintain resting Ca2+ levels (18).
The Na+–K+ ATPase comprises α- and β-subunits. The α-subunit contains the binding sites for ATP, Na+, K+, cardiac glycosides, and is thus largely responsible for the catalytic, transport, and pharmacologic characteristics of the ATPase. The smaller β-subunit modulates the transport characteristics of the ATPase and plays an important role in its proper membrane insertion (18). There are three Na+–K+ pump α-subunits in the heart that display similar ion affinities and ATPase activity, but different affinities for cardiac glycosides and distinct subcellular localizations. In the rodent heart, the low digitalis affinity α1 isoform predominates through all phases of development, while there is a postnatal transition from the neonatal α3 isoform to the adult α2 isoform that occurs within the second week of postnatal life (19). However, in humans the α1 isoform has high affinity for cardiac glycosides. In the adult human heart, three different isoforms of the Na+/K+-ATPase, α1β1, α2β1, and α3β1, are expressed with different affinities for different cardiac glycosides (20). To date, there have been no human studies characterizing developmental changes in Na+/K+-ATPase isoform expression.
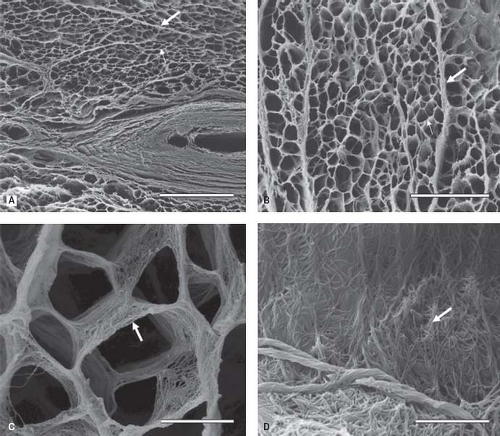 Figure 4.2 Connective tissues of the human heart (transverse section). A: The collagen network around cardiomyocytes and small vessels is observed. Bar = 200 μm. (thick arrow: perimysium; thin arrow: endomysium). B: The interstitial connective tissue consisting of perimysial and endomysial components presents a honeycomb shape. The perimysium (thick arrow) surrounds groups of cardiomyocytes, and the endomysium (thin arrow) surrounds each cardiomyocyte. Bar = 300 μm. C: The endomysium (arrow) supports and connects individual cardiomyocyte fascicles. Magnification ×3000, bar = 10 μm. D: At higher magnification, collagen fibers show interconnections on the surface of cardiomyocytes. Thin collagen strands are probably collagen III (arrow). Magnification ×10,000, Bar = 3 mm. (Kanzaki Y, Terasaki F, Okabe M, et al. Three-dimensional architecture of cardiomyocytes and connective tissue in human heart revealed by scanning electron microscopy. Circulation 2010;122:1973–1974, with permission.) |
Na+/K+-ATPase activity indirectly regulates other plasma membrane transporters that require an inward Na+ gradient. The cardiac Na+/Ca2+ exchanger (NCX1) is the major Ca2+ extrusion mechanism in cardiac myocytes by utilizing the Na+ electrochemical gradient to mediate the electrogenic countertransport of three Na+ ions for one Ca2+ ion across the sarcolemmal membrane (21). This exchanger is bidirectional and capable of moving Ca2+ in either direction across the sarcolemma.
The plasma membrane also contains a high-affinity, low-capacity Ca2+-ATPase that extrudes Ca2+ from the cell in an ATP-dependent process. This pump functions as a fine-tuner of cell Ca2+, lowering it to the submicromolar level (22), thereby maintaining low amounts of intracellular Ca2+ during basal conditions.
Na+ Channels, Ca2+ Channels, K+ Channels: The Cardiac Action Potential
Under resting conditions, the membrane potential (Vm) is set by the intracellular and extracellular concentrations of Na+ and K+, and the conductance properties of ion channels on the plasma membrane (Fig. 4.3). This equilibrium potential (voltage at which net flow is zero) for a single ion is described by the Nernst equation. In ventricular myocytes, the resting Vm (∼−86 mV) is close to that predicted by the Nernst equations for K+ (23). In order to elicit an action potential, any electrical stimulus must depolarize the membrane to a threshold value (∼−65 mV) necessary for activation of voltage-sensitive Na+ channels.
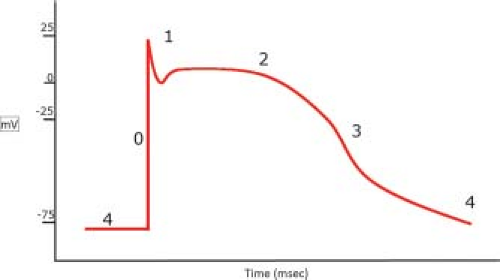 Figure 4.3 Schematic representation of the cardiac action potential. |
Phase 0: The rapid depolarization phase and action potential amplitude is due to Na+ entry through voltage-sensitive Na+ channels encoded by the SCN5A gene. Once open, these channels rapidly inactivate at higher Vm and undergo a refractory period in which the channels become unresponsive to any further stimulation. This voltage-dependent activation and inactivation of the channel are important clinically since changes in its expression or gating properties can affect action potential amplitude and durations, leading to arrhythmias (23).
Phase 1: Early repolarization. The brief repolarization phase results in the notch between the end of the upstroke and the beginning of the plateau phase. This transient outward current is largely due to activation of K+ channels. There are numerous K+ channels present in the sarcolemma that are characterized by their gating properties and substances that regulate their opening (e.g., KAch, KCa) and closing (e.g., KATP) (24). The K+ channels involved in this phase are outward rectifier Kv channels (i.e., pass current better in the outward direction).
Phase 2: The plateau phase. Ca2+ enters the cell through voltage-gated L-type Ca2+ channels, which are composed of two subunits (α and α1) that form the ion pore. These channels are regulated by membrane potential and the inward Ca2+ concentration gradient. In the adult heart, the majority of Ca2+ influx occurs through this channel, although fetal myocytes also express T-type Ca2+ channels that may contribute to E-C coupling. During this phase, delayed, outward rectifier K+ channels begin to open and the positive Vm drives K+ efflux from the cell.
Phase 3: The repolarization phase. K+ efflux through outward rectifier K+ channels and Ca2+-activated K+ channels (KCa or BK channels) dominate this repolarization phase. The KCNQ1 and KCNE1 genes encode subunits of the channels that conduct the slow component of delayed rectifier K+ current (25). Mutations in the KCNQ1 gene encoding a delayed rectifier K+ channel are also associated with long Q-T syndrome (26). Homozygous mutation KCNQ1 and KCNE1 result in Jervell and Lange-Nielson syndrome, characterized by severe QT interval prolongation and high risk of sudden cardiac death (25). On the other hand, activation of the KCa channel by Ca2+ explains why the QT interval is diminished by cardiac glycosides (23). Expression of these channels changes over the course of cardiac development, which likely influence changes in the action potential duration and repolarization (27).
Phase 4: Restoration of ionic concentrations: This phase is largely driven by K+ flux through inwardly rectifying K+ channels. The Na+/K+ ATPase also maintains the resting membrane potential at −90 mV.
Sarcoplasmic Reticulum
The sarcoplasmic reticulum (SR) is a network of tubular membranes that surround the myofibrils and are involved in the regulation of Ca2+ concentrations (Fig. 4.4). The SR is composed of two distinct regions that are involved in Ca2+ regulation: the junctional SR regulates Ca2+
storage and release, while the longitudinal SR regulates Ca2+ uptake. Release of SR Ca2+ is a tightly regulated process and proper functioning of the SR is imperative for normal heart function. The SR contains ryanodine Ca2+ release channels that play a primary role in E-C coupling (28) (see below). The cardiac SR Ca2+-ATPase (SERCA2a) pumps Ca2+ into the SR against its concentration gradient. SERCA2a is the main player in restoring diastolic Ca2+ levels and terminating Ca2+-dependent force activation. The function of SERCA2a can be modulated by several indirect and direct factors. The most predominant indirect mechanism is inhibition by phospholamban. Phosphorylation of phospholamban via β-adrenergic stimulation and enhanced cAMP-dependent protein kinase-A (PKA) activity relieves SERCA2a inhibition (29). SERCA2a is also under the direct control of Ca2+/calmodulin-dependent protein kinase II, which has been shown to phosphorylate SERCA2a and enhance its Ca2+ transport capacity (29). During development, the SR content of the heart changes wherein the immature heart SR content is dramatically less (30,31) than in the mature heart (32).
storage and release, while the longitudinal SR regulates Ca2+ uptake. Release of SR Ca2+ is a tightly regulated process and proper functioning of the SR is imperative for normal heart function. The SR contains ryanodine Ca2+ release channels that play a primary role in E-C coupling (28) (see below). The cardiac SR Ca2+-ATPase (SERCA2a) pumps Ca2+ into the SR against its concentration gradient. SERCA2a is the main player in restoring diastolic Ca2+ levels and terminating Ca2+-dependent force activation. The function of SERCA2a can be modulated by several indirect and direct factors. The most predominant indirect mechanism is inhibition by phospholamban. Phosphorylation of phospholamban via β-adrenergic stimulation and enhanced cAMP-dependent protein kinase-A (PKA) activity relieves SERCA2a inhibition (29). SERCA2a is also under the direct control of Ca2+/calmodulin-dependent protein kinase II, which has been shown to phosphorylate SERCA2a and enhance its Ca2+ transport capacity (29). During development, the SR content of the heart changes wherein the immature heart SR content is dramatically less (30,31) than in the mature heart (32).
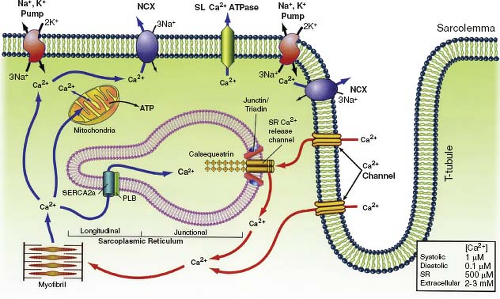 Figure 4.4 Pumps and channels regulate Ca2+ signaling, the key modulator of contraction and relaxation. Following depolarization, Ca2+ enters the cells through the sarcolemma and causes Ca2+-induced Ca2+ release from the sarcoplasmic reticulum, initiating contraction. Relaxation occurs when most of the Ca2+ is resequestered into the SR. SR, sarcoplasmic reticulum; SL, sarcolemma; PLB, phospholamban; NCX, Na–Ca exchanger. |
T-Tubules
The system of T-tubules extends transversely into the center of the myocyte and envelops the myofibril at the level of the Z-disc, forming couplings with the SR. This allows transmission of the action potential to the cellular interior, leading to rapid activation of the cell. In humans, T-tubules are noted at 30 weeks’ gestation (33). The development of T-tubule networks appears variable, with myocytes from animals that are well developed at birth having well-developed t-tubule systems and those from less well-developed neonates lacking mature t-tubule networks. These variations in t-tubule development may account for the variability in E-C coupling between mature and immature heart cells.
Sarcomere
The sarcomere (Fig. 4.5) is the functional unit of heart muscle and is composed of interdigitating thick (myosin) and thin (actin) filaments, structural components, and regulatory proteins. Each sarcomere is bordered at each end by a Z-disc of ∼0.1 μm length. The Z-disc bisects the I-bands of adjacent sarcomeres, which contains thin filaments, troponin (Tn), and tropomyosin (34). The A-band is a region of overlapping thick and thin filaments, while the H-zone (the center is termed the M-line) contains thick filaments linked to titin and myosin-binding proteins.
Z-Disc
The Z-discs demarcate individual sarcomeres and directly interact with all myofilament proteins except myosin. α-Actinin is the primary protein that makes up the backbone of the Z-disc and cross-links the ends of actin to adjacent sarcomeres (35). Proteins in the Z-disc also bind the intermediate filament desmin to link the sarcomere to the intercalated disc and costameres at the plasma membrane (see following section). This unique structure allows the integration of signals from the cytoskeleton, ECM, and those induced by mechanical stress (35).
Thin Filaments
The thin filaments are composed of a double helix of two F-actin filaments. There are two isoforms of actin, skeletal, and cardiac α-actin, that differ by four amino acids. Early in human heart development both α-actin isoforms are expressed (36). As development progresses, however, the content of skeletal α-actin is decreased, while the cardiac isoform is increased; however, the mechanisms of this isoform switch have not been delineated. The Tn complex is composed of three proteins: TnT, I, and C. TnT facilitates binding of the Tn complex to tropomyosin. Tropomyosin is evident in both α and β isoforms that wind together to form a coil. TnI is a strong inhibitor of actin–myosin interactions, binding to TnC during systole and actin during diastole (Fig. 4.6) (37). TnI also has skeletal and cardiac forms, and both are expressed in immature human hearts (38). During development of the heart, the skeletal isoform is changed to the cardiac isoform, which may contribute to the changes in cardiac function during development (39). TnC binds Ca2+, which results in its conformational change that causes TnT to lift tropomyosin and TnI away from the ATP reactive site to permit actin and myosin interaction leading to contraction.
The distribution of thin filament regulatory proteins is altered during heart development and likely contributes to developmental changes in myocardial contraction. TnI exists in two isoforms, cardiac TnI and slow skeletal TnI. Slow skeletal TnI is the major isoform during development, and a switch occurs at birth to cardiac TnI (40). This switch is postulated to account for the decrease in calcium sensitivity and pH responsiveness found in the adult heart as compared to the neonatal heart (40,41). TnT has multiple isoforms. Studies in the rat heart demonstrated that TnT1 is the major isoform in the fetal heart with a switch to TnT2 in the neonate (42). However, in human studies, TnT1 is the predominant isoform in both the fetus and adult while TnT2 is only found in the fetal heart (43). Interestingly, TnC does not seem to change expression
during development. Tropomyosin also has two major isoforms, α and β. In the adult heart, these two forms are present at approximately equal levels, but during development, β-tropomyosin has lower expression levels. During late fetal development, expression of this β-isoform increases to adult levels, and this is postulated to be inversely correlated with heart rate (23).
during development. Tropomyosin also has two major isoforms, α and β. In the adult heart, these two forms are present at approximately equal levels, but during development, β-tropomyosin has lower expression levels. During late fetal development, expression of this β-isoform increases to adult levels, and this is postulated to be inversely correlated with heart rate (23).
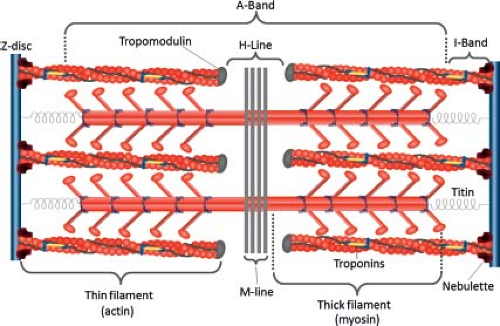 Figure 4.5 Protein composition of the sarcomere. Titin, extending from the Z band to the thick filament, connects to myosin by myosin-binding protein C. Several proteins support the thin filaments, including nebulette (connecting the thin filaments to the Z band) and tropomodulin (linked to the ends of thin filaments). (Adapted from Clowes C, Boylan MG, Ridge LA, Barnes E, Wright JA, Hentges KE. The functional diversity of essential genes required for mammalian cardiac development. Genesis 2014;52:713–737. (186).) |
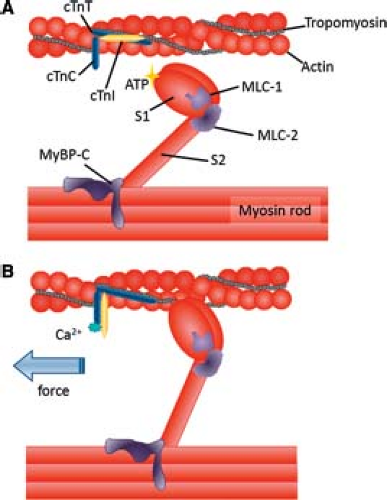 Figure 4.6 Schematic illustration of the sarcomere during diastole (A) and systole (B). Thin filament proteins include tropomyosin and the troponin complex: Troponin C (TnC), Troponin T (TnT), and TroponinI (TnI). The thick filament comprises myosin and myosin-binding proteins such as cardiac myosin–binding protein-C (cMyBP-C). Myosin heavy chain is an asymmetric molecule with a globular head (S1) and a rod-like tail separated by a hinge region (S2). The S1 region contains both the ATP hydrolysis domain and the actin-binding domain. The myosin head is associated with an essential light chain (MLC-1) and a regulatory light chain (MLC-2). During diastole, Tn-I exerts an inhibitory effect, preventing actin–myosin interaction. B: Systole. Ca2+ binding to Tn-C induces a conformational change in the troponin–tropomyosin complex. Movement of tropomyosin exposes a myosin-binding site on actin allowing crossbridge formation to take place. (Adapted from Hamdani N, Kooij V, van Dijk S, et al. Sarcomeric dysfunction in heart failure. Cardiovasc Res 2008;77:649–658 with permission from Oxford University Press.) |
Thick Filaments
Myosin is the most abundant protein in the myocyte, with two heavy chains and four light chains. The two heavy chains of myosin wind together to form the thick filament, with the myosin heads projecting up from the long axis of the thick filament to form crossbridges. In the presence of Ca2+, the myosin heads bind to actin to initiate contraction. In the first step, the myosin head, due to its intrinsic ATPase activity, becomes activated by ATP hydrolysis, allowing the head to attach to actin and form a crossbridge. The interaction elicits a conformational change in the myosin head that pulls the actin filament inward (“power stroke”). ADP is then released and a new molecule of ATP binds to the myosin head, causing it to release the actin filament. If intercellular Ca2+ remains elevated, myosin will undergo another crossbridge cycle.
In cardiac muscle, the relative expression of the two isoforms of myosin heavy chain (MHC)-α and β, depends upon species, heart size, and heart rate. Detailed expression studies in mouse embryos demonstrate that the predominant isoform in developing ventricles is MHC-β while MHC-α expression is localized to the developing atria. MHC-β is found as early as E7.5 in the mouse embryonic heart (44). In early postnatal life in the rodent, there is a switch from MHC-β to MHC-α in the ventricles. Compared with MHC-β in the same species, MHC-α displays faster sarcomeric shortening velocity, higher power production, and greater rate of force development, but at the expense of increased oxygen demand (45).
In human ventricles, MHC-β is the predominant form during all stages of life (46). As discussed in later chapters, mutations in MHC-β are a prevalent cause of hypertrophic cardiomyopathy. MHC-α is found in only very small amounts in the human ventricle, but mutations in this protein are linked to both hypertrophic and dilated cardiomyopathy (47). Expression of the myosin light chain (MLC) does not appear restricted in the early to mid-fetal stages of development, but by late fetal life, MLC1A is restricted to the atria and MLC1V to the ventricles. This expression is maintained in adulthood.
Cardiac myosin binding protein-C (cMyBP-C) is a thick-filament accessory protein localized to the A-band of the sarcomere and binds to myosin, actin, and titin. The interaction of its C-terminal domains with myosin and titin are thought to stabilize and organize the thick myofilament and truncations in this region lead to dilated cardiomyopathy. Unphosphorylated cMyBP-C limits crossbridge formation by constraining the myosin head; phosphorylation relieves this constraint by increasing the proximity of myosin heads to actin, leading to an increase in the probability of actin–myosin. Although the exact mechanism is unclear, phosphorylation of cMyBP-C appears to accelerate crossbridge kinetics to increase force development and subsequent relaxation. Mutations in cMyBP-C are linked to LV dilation, cardiac hypertrophy, and contractile dysfunction, depending upon whether the mutation leads to cMyBP-C truncation or mutations that affect actin–myosin interactions (48).
Titin
Titin is the largest protein known, spanning half of the sarcomere from Z-disc to M-band (Fig. 4.5). Titin’s remarkable elasticity is due to its I-band domain that comprises three sequence elements that develop passive tension as the sarcomere is stretched. These three elements comprise distal and proximal immunoglobulin (Ig) domain, a unique N2B segment (short molecular spring), and the PEVK segment (named for its rich composition of proline (P), glutamate (E), valine (V), and lysine (K) residues). Tension generated by these elements is responsible for pressure generated during diastole and accounts for approximately 80% of passive stiffness at physiologic sarcomere lengths (49). Titin’s passive force is modulated during development and in response to pathologic changes in hemodynamic load. This occurs primarily by differential splicing primarily in the Ig and PEVK segments, which leads to three different isoform classes with variable compliance (50). The stiffer N2B isoform is the shortest of these and is approximately 3.0 MDa. The second class is the more compliant N2BA isoform (long molecular spring), which is approximately 3.3 to 3.5 MDa, and contains additional PEVK segments and a varying number of additional Ig domains.
The relationship of titin to cardiac development and disease is complex. A dramatic isoform switch occurs during cardiac development in mammals, from fetal N2BA titin expressed before birth to a mix of smaller N2BA/N2B isoforms found postnatally. Adult rat hearts almost exclusively have N2B titin isoform (51,52). These changes in titin isoform composition during heart development affect myofibrillar extensibility and passive force generation, and alter cardiac stiffness (53). In humans, N2BA/N2B ratios vary with disease from an increased ratio in dilated cardiomyopathy (more compliant) (49,54,55,56) to decreased ratios in aortic stenosis (less compliant) (49,55,56,57).
Mitochondria and Metabolism
During development, the heart requires an abundant amount of energy to grow, differentiate, and mature. Cellular mitochondria are the key “powerhouses” of energy production, generating abundant amounts of ATP for use in cellular processes. Relative to other organs, the heart contains a high number of mitochondria to accommodate this energy need.
Mitochondria are rectangular shaped with an inner membrane that folds into numerous compartments termed cristae. The cristae greatly expand the intracellular surface area to accommodate membrane-bound enzymes of the electron transport chain responsible for generating ATP in the inner matrix. Adult myocytes contain two distinct types of mitochondria: Subsarcolemmal mitochondria located beneath the plasma membrane, and interfibrillar mitochondria located between myofibrils in parallel rows and are anchored by the tubulin cytoskeleton (58). The number, size, and volume of mitochondria increase during development coupled with lengthening of the cristae. During the early postnatal period, mitochondria are often randomly clustered throughout the cytoplasm, but start to align along the β-tubulin bundles, adjacent to myofibrils, the plasma membrane, and the SR (58).
Cellular metabolism and energetics undergo major shifts during the switch from fetal life to adulthood. The fetal heart uses predominately glucose and glycolysis to generate ATP. At birth, the increase in LV cardiac output increases energy demands, and along with the postnatal increase in oxygen tension, drives mitochondrial biogenesis and oxidative phosphorylation (59). At this stage, the heart switches to β-oxidation of fatty acids that supply reducing equivalents to the mitochondrial electron transport chain as its primary substrates for metabolism (60). At the cellular level, these changes are coordinated by a specialized transcriptional circuit regulated by peroxisome proliferator–activated receptor-γ coactivator-1 (PGC-1) (61). The adult heart utilizes all classes of energy substrates, including carbohydrates, lipids, amino acids, and ketone bodies for mitochondrial ATP.
Cell–Extracellular Matrix and Cell–Cell Interactions
Cardiac Myocyte–Extracellular Matrix Interactions
Cardiac myocyte contraction causes cellular deformation and shortening (Fig. 4.7). During this process, the contractile machinery must remain connected to the surrounding ECM; otherwise, contraction would be improperly transmitted which would increase the risk of membrane damage (62). Cells maintain strong connection to the ECM through two specialized complexes, costameres and dystrophin glycoprotein complexes. Adhesions between myocytes and the ECM reorganize during development to stabilize tissue during myofibrillogenesis and disassemble as systolic stresses are transmitted intercellularly via mature intercalated discs (63). Cell–ECM adhesions are active mechanosensing units responsible for inducing the expression and phosphorylation of an array of protein kinases, such as phosphoinositide-3-kinase/AKT p38 mitogen-activated protein kinase important to myocyte maturation (64). These cell–ECM adhesions are in part responsible for sensing the stiffness of the surrounding ECM and transduction of these signals to regulate cardiomyogenesis.
Costameres
Costameres are complex protein structures containing integrins that form a physical connection between the ECM and outer Z-discs of underlying sarcomeres. This complex acts to transmit the contractile forces generated by the myofilaments directly to the ECM. Integrins are heterodimeric proteins consisting of single α- and β-subunits. Cardiac myocytes express multiple α-subunits (α1-, α3-, α5-, α6-, α9– and αv-subunits) whereas the predominant β-subunit is β1A and, to a lesser extent, β1D (65). Different combinations of these subunits allow integrins to bind to specific peptide sequences in basal membrane proteins (e.g., collagen IV and laminin) and to Type I and III collagens
and fibronectin in the ECM. The α-subunit directly binds to the matrix proteins, while the cytoplasmic tail of β-integrin interacts with several cytoskeletal proteins (vinculin, talin, filamin) that directly link to α-actinin at the Z-disc and cytoskeletal actin. This spatial organization of integrin-associated costameres allows them to act as key mechanical sensors that transduce changes in mechanical forces to cellular signaling cascades that mediate sarcomere assembly, gene expression, cell migration, and survival. This type of “outside-in” signaling is the primary mechanism that regulates cardiac myocyte growth in response to changes in hemodynamic load (65).
and fibronectin in the ECM. The α-subunit directly binds to the matrix proteins, while the cytoplasmic tail of β-integrin interacts with several cytoskeletal proteins (vinculin, talin, filamin) that directly link to α-actinin at the Z-disc and cytoskeletal actin. This spatial organization of integrin-associated costameres allows them to act as key mechanical sensors that transduce changes in mechanical forces to cellular signaling cascades that mediate sarcomere assembly, gene expression, cell migration, and survival. This type of “outside-in” signaling is the primary mechanism that regulates cardiac myocyte growth in response to changes in hemodynamic load (65).
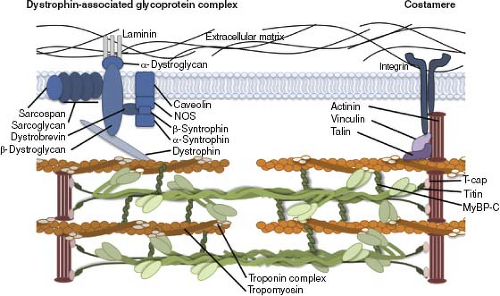 Figure 4.7 Extracellular matrix–cardiac myocyte interactions. Attachment to the ECM is mediated by costameres composed of the dystroglycan–glycoprotein complex and the integrin complex. Force transduction and intracellular signaling are coordinated through the costamere. The unique roles of each of these proteins are critical to appropriate structure and function of the heart. T-cap, titin cap; MyBP-C, myosin-binding protein C. (From Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol 2011;194:355–365, with permission.) |
Costameres play a key role in sarcomere assembly. Sustained changes in myocardial wall stress and strain result in myocyte growth or atrophy. For example, cells elongate in response to sustained diastolic strain by adding sarcomeres in series, while they thicken in response to increased systolic stress by adding sarcomeres in parallel (66). The precise mechanisms by which new sarcomeres are added to the myofilament are unknown. Russell et al. performed a series of gene overexpression or deletion studies in neonatal cardiac myocytes which suggests that integrins activate signaling cascades (e.g., protein kinase E-C) that “loosen” the Z-disc, weakening actin binding and stimulating actin capping and myofilament formation (66,67,68).
The molecular composition of integrins and the ECM changes throughout development, suggesting that these two factors may contribute to myocardial morphogenesis. One example is the coordinated regulation of fibronectin and β-integrins. The levels of fibronectin and β1-integrin are elevated in the developing myocardium, which allow myocytes to spread and proliferate, but in adult cells that have lower levels of these proteins, myocytes only attach weakly to fibronectin. Both neonatal and adult cells bind strongly to laminin and collagen IV in the basal lamina (63).
Dystrophin-Associated Glycoprotein Complexes
The dystrophin-associated glycoprotein complex provides a strong mechanical link from the cardiac cytoskeleton to the ECM and appears to colocalize with costameres (Fig. 4.7). Components of this complex include dystrophin, dystroglycan, sarcoglycans, and dystrobrevins. Mutations in these proteins result in plasma membrane damage in striated muscle as seen in muscular dystrophy–associated cardiomyopathies. Loss of normal dystrophin function in the heart produces four-chamber dilation, reduction in left ventricular function, and arrhythmias (69). Since dystrophin complex–related cardiomyopathies will be covered in great detail in a later chapter, this section will be restricted to an overview of dystrophin complex organization and function, as well as functional classes of mutations.
Dystrophin is a rigid cytoplasmic protein that anchors dystroglycan to the cytoskeletal, filamentous actin (70). Removal of dystrophin from the dystrophin glycoprotein complex makes this complex unstable and leads to its loss from the plasma membrane, rendering the cell susceptible to damage from contraction (71). Specifically, the loss of membrane integrity allows Ca2+ entry into the cell, which in turn activates Ca2+-sensitive proteases, leading to cellular degradation and release of cardiac myocyte proteins such as creatine kinase into the bloodstream (72,73). In cardiac myocytes, mutations in dystrophin also affect the function of stretch-activated ion channels, which normally open in response to stretch during ventricular filling (72).
Heterodimeric dystroglycan is a protein central to the dystrophin glycoprotein complex that spans the sarcolemma and binds to laminin in the surrounding basal lamina through its α-dystroglycan subunit and to dystrophin through the cytoplasmic, carboxy-terminus of its β-dystroglycan subunit (74,75). Sarcoglycans are transmembrane glycoprotein complexes comprising six isoforms (α, β, δ, ε, γ, ζ) that are thought to stabilize the interactions between α and β dystroglycans. Dystrobrevin binds to dystrophin and the sarcoglyan complexes and also plays an important role as a structural scaffold linking the dystrophin glycoprotein complex to intermediate filaments (76). The intermediate filaments, in turn, encircle the Z-discs of each myofibril. Mutations in dystrobrevin have been associated with left ventricular noncompaction, a cardiomyopathy characterized by a pattern of prominent trabecular meshwork and deep trabecular recesses (77).
ECM and Mechanobiology
Both fibroblasts and myocytes are mechanically sensitive to external physical forces (Fig. 4.8). The cytoskeleton creates direct mechanical links to the ECM via transmembrane integrin receptors and linker proteins. These linkages cluster to form focal adhesions and provide a route through which external physical forces are transduced into intracellular process. When a cell experiences increased extracellular resistance, it increases actin polymerization in order to create an internal cytoskeletal tension proportional to the force exerted by the ECM. This dynamic interplay between the ECM and the cytoskeleton dictates many aspects of cell behavior.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


