Fig 1.
A 13-year-old girl with juvenile idiopathic arthritis. Axial lung window computer tomography (CT) shows bronchiectasis in the posterior lower lobes. Right pleural effusion is also seen
Some specific lung abnormalities are associated with a specific collagen vascular disease. For example, lipoid pneumonia, caused by deposition of cholesterol granulomas in the interstitium and alveoli, is associated with JIA, which may be related to an underlying macrophage activation syndrome that is often present in JIA [5]. Alveolar hemorrhage, which is more common in children than in adults, may present in pediatric patients with SLE and decreased hematocrit level (Fig. 2). The early and accurate diagnosis of alveolar hemorrhage in SLE patients is essential because it can be potentially treated successfully with steroids and cytotoxic agents. Furthermore, “shrinking lung syndrome,” which describes a progressive decrease in lung volumes and is sometimes seen in patients with SLE, can be detected on imaging studies when there is a progressive elevation of the diaphragm despite attempted full inspiration [6]. Although the underlying etiology of “shrinking lung syndrome” is not known, a combination of pleural restriction due to recurrent pleural inflammation and pulmonary fibrosis, and weakness of the diaphragm and chest wall musculature may be responsible.
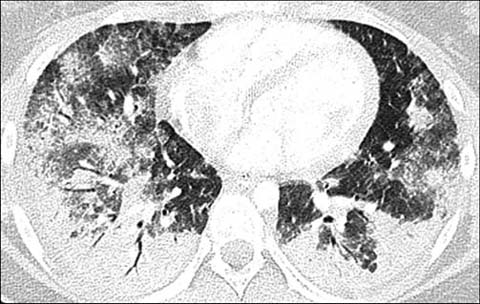
Fig 2.
A 15-year-old girl with a known diagnosis of systemic lupus erythematosus who presented with shortness of breath, desaturation, and decreased hematocrit. Axial lung window CT shows multifocal airspace opacifications compatible with pulmonary hemorrhage. A subsequently performed bronchoalveolar lavage confirmed the diagnosis
Immunodeficiencies
Defects in one or more of the essential components of the immune system can result in an increased risk of developing thoracic abnormalities in pediatric patients [3]. When assessing the chest of children with an immunodeficiency, the radiologist’s role includes: (1) detecting characteristic imaging features of a specific immunodeficiency, (2) evaluating infections, and (3) detecting malignancies that can occur as a complication of certain immunodeficiencies. Because of the large number of immunodeficiencies, only the disorders that are the most common or have the most characteristic thoracic manifestations are discussed in the following.
Predominantly Antibody Deficiencies
The predominantly antibody deficiencies are characterized by a decreased ability to produce immunoglobulins from B-cells. The predominantly antibody deficiencies predispose affected patients to recurrent respiratory tract infection with encapsulated bacteria such as Streptococcus and Haemophilus influenzae. Recurrent infections from these organisms can lead to permanent lung damage such as irreversible bronchiectasis. The four most commonly encountered predominantly antibody deficiencies are IgA deficiency, X-linked agammaglobulinemia, common variable immunodeficiency disorder (CVID), and hyper-IgM syndrome.
In children with IgA deficiency, which is the most common primary immunodeficiency syndrome, pyogenic sinopulmonary infections are the most frequent finding [7]. X-linked agammaglobulinemia most often results from a mutation in the X-linked gene encoding Bruton tyrosine kinase (Fig. 3). Following recurrent pulmonary infections from Streptococcus, H. influenzae, and Mycoplasma, bronchiectasis in the lower lobes is often seen. Diminutive adenoid tissue on the lateral airway radiograph can be helpful clue for diagnosing X-linked agammaglobulinemia in affected children. CVID is described as “variable” because of the different clinical courses of different patients, rather than to changing manifestations over time in a single patient. CVID is caused by the failed terminal differentiation of B-cells to plasma cells, resulting in hypogammaglobulinemia. Approximately 75% of affected patients develop a lung disease characterized by bronchiectasis, bronchial wall thickening, and endobronchial mucus plugging closely resembling cystic fibrosis [8] (Fig. 4). Splenomegaly is seen in approximately 25% of patients with CVID [7]. Affected patients also have an increased risk for developing autoimmune cytopenia, lymphoproliferative disease, lymphoma, and gastric cancer. In addition, patients with CVID may develop granulomatous lymphocytic-interstitial lung disease (GLILD). On imaging studies, GLILD is characterized by a non-caseating granulomatous and lymphoproliferative histologic pattern often seen as pulmonary nodules, consolidation, and ground-glass or reticular opacities [9] (Fig. 5). Children affected with hyper-IgM syndrome may present with recurrent respiratory and gastrointestinal tract infections by encapsulated bacteria, viruses, fungi, and parasites [10]. An increased risk of developing Pneumocystis jirovecii pneumonia (PCP) and hepatocellular carcinoma has been also reported in patients with hyper-IgM syndrome.
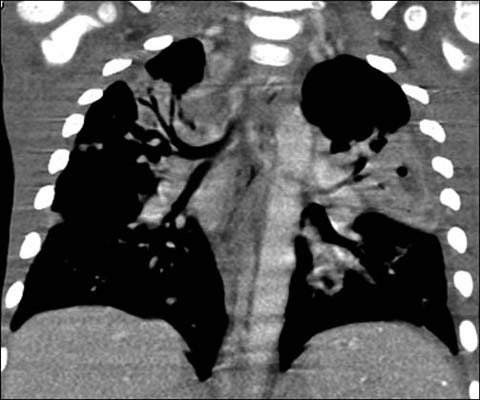
Fig 3.
A 3-year-old boy with X-linked agammaglobulinemia. Enhanced coronal reformatted CT shows multi-focal pneumonia from Streptococcus infection
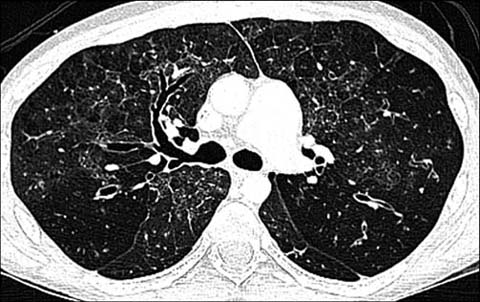
Fig 4.
A 16-year-old boy with common variable immunodeficiency disorder. Axial lung window CT shows bronchial wall thickening and bronchiectasis closely resembling cystic fibrosis
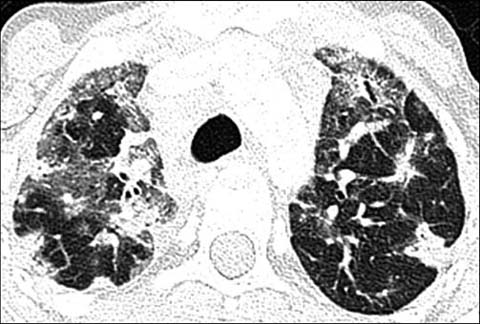
Fig 5.
A 17-year old girl with common variable immunodeficiency disorder. Axial lung window CT shows nodules as well as ground-glass and subtle reticular opacities in both lungs, compatible with granulomatous lymphocytic-interstitial lung disease (GLILD)
Combined T-cell and B-cell Immunodeficiencies
Pediatric patients with combined T-cell and B-cell immunodeficiencies are at increased risk of developing infections from the same pathogens that afflict those with antibody deficiencies. In addition, opportunistic infections from Mycobacteria, viruses, Pneumocystis, and other fungi can occur [11]. The two most common combined T-cell and B-cell immunodeficiencies are severe combined immunodeficiency (SCID) and combined immunodeficiency (CID).
In patients with SCID, the most severe form of the primary immunodeficiencies, recurrent, persistent, or opportunistic respiratory infections along with thrush, dermatitis, chronic diarrhea, or failure to thrive begin in early infancy. Without hematopoietic stem cell transplantation, gene therapy or enzyme replacement therapy, the disease is almost always fatal in the first two years of life [12]. In patients with CID, particularly with signal transducer and activator of transcription 5B (STAT5b) deficiency, the chronic lung disease in the form of lymphocytic interstitial pneumonitis can result in death from respiratory failure [13].
Well-Defined Syndromes Associated with Immunodeficiency
The five most common well-defined syndromes associated with immunodeficiency in the pediatric population are: (1) DiGeorge syndrome, (2) Wiscott-Aldrich syndrome, (3) hyper-IgE syndrome, (4) dyskeratosis congenita, and (5) ataxia-telangiectasia.
DiGeorge syndrome, also known as a velocardiofacial syndrome, is characterized by developmental malformation of the third and fourth pharyngeal pouches. This leads to distinctive facial features (hypertelorism, saddle nose, short philtrum, low-set ears, cleft palate), conotruncal malformations (including tetralogy of Fallot, interrupted aortic arch, or truncus arteriosus), thymic hypoplasia, and parathyroid hypoplasia [14]. Typical thoracic imaging findings that may be present in pediatric patients with DiGeorge syndrome are a narrowed mediastinum related to a small thymus, an abnormal cardiac silhouette related to a conotruncal malformation, and recurrent pulmonary infections. Large-airway abnormality, such as tracheobronchomalacia, and osseous anomalies involving the ribs or vertebrae may be also present. In Wiscott-Aldrich syndrome, pulmonary infections with encapsulated Streptococcus pneumoniae and Pneumocystis are often seen [15]. In patients with hyper-IgE syndrome, recurrent pneumonia, lymphadenopathy, post-infectious pneumatoceles, and bronchiectasis are typically features on imaging studies. In addition, some patients have osseous findings, including osteoporosis, fractures, and scoliosis [16]. Children with dyskeratosis congenita, which is a progressive multisystemic disorder, characteristically present with rapidly progressing pulmonary fibrosis after hematopoietic stem cell transplantation [17] (Fig. 6). Ataxia-telangiectasia is due to underlying autosomal recessive mutations in the ATM gene, which encodes a DNA damage response protein. Affected pediatric patients are highly susceptible to DNA damage from ionizing radiation and the subsequent development of cancer, including lymphoma, leukemia, and leiomyosarcoma. Thus, in these patients, every effort should be made to limit imaging using ionizing radiation. Characteristic thoracic imaging findings include a small thymus, recurrent pneumonia (from S. aureus, H. influenzae, and S. pneumoniae), bronchiectasis, and lymphadenopathy [18] (Fig. 7). In addition, a chronic interstitial lung disease unique to ataxia-telangiectasia (ATILD) may occur. On imaging studies it is characterized by pulmonary fibrosis presenting as interstitial and pleural thickening.
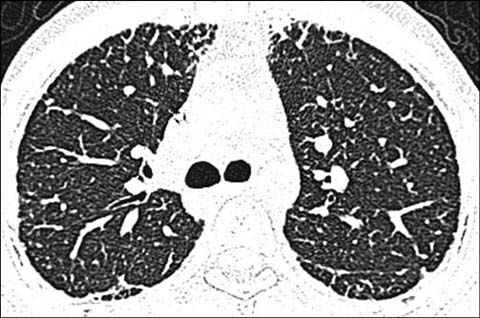
Fig 6.
A 15-year-old girl with dyskeratosis congenital status post hematopoietic stem cell transplantation. Axial lung window CT shows fibrotic lung changes in the peripheral portion of the lungs
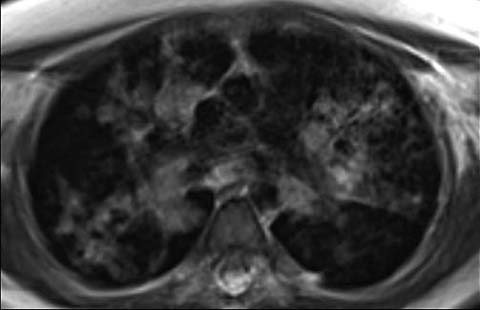
Fig 7.
An 11-year-old girl with ataxia-telangiectasis who underwent MRI of the lungs due to increased susceptibility to DNA damage from ionizing radiation associated with CT. Axial MRI shows multiple nodular and airspace opacities from histoplasmosis infection. The patient also later developed lymphoma (not shown)
Congenital Defects of Phagocyte Number and/or Function
The most commonly encountered immunodeficiency syndrome related to congenital defects of phagocytes in children is chronic granulomatous disease (CGD). Affected children typically present with granuloma and abscess formation from catalase-positive S. aureus, fungi, or atypical mycobacteria (Fig. 8). Although CGD can affect any organs, the lungs are the most common location of infection [19]. Depending on the infectious organism and disease chronicity, nodules, ground-glass opacities, consolidation, septal thickening, and cysts can be seen in the early phase. Later, recurrent and long-standing lung infections can lead to permanent damage, such as pulmonary fibrosis, honey-combing, and pulmonary hypertension. Extrapulmonary thoracic manifestations include suppurative lymphadenopathy, pleuritis, empyema, and rib or vertebral osteomyelitis.
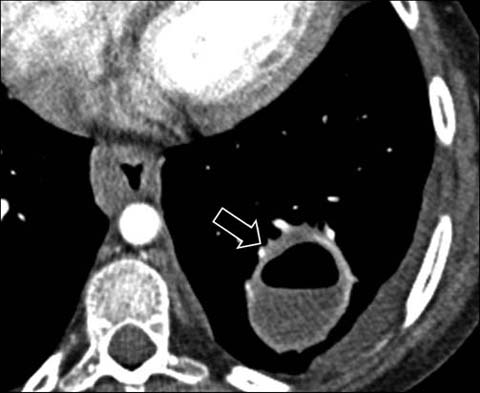
Fig 8.
A 14-year-old girl with chronic granulomatous disease. Enhanced axial CT shows a large lung abscess (arrow) from S. aureus infection. Also note the esophageal thickening due to chronic infective esophagitis
Acquired Immunodeficiency
The two most commonly encountered acquired immunodeficiencies in the pediatric population are human immunodeficiency syndrome (HIV) and neutropenia due to chemotherapy for cancer or allogeneic hematopoietic stem cell transplantation. The lungs are the organ most commonly affected by infections in children with acquired immunodeficiency.
Many bacterial and viral infections have diverse nonspecific radiographic appearances in children with HIV Pulmonary infection with PCP has more characteristic imaging findings; these include bilateral diffuse or patchy ground-glass or reticulonodular opacities with perihilar to peripheral progression [20]. In addition, pulmonary cysts, which can rupture and result in pneumothorax or pneumomediastinum, may also present on imaging studies in children with HIV Lymphocytic interstitial pneumonitis (LIP) is a form of pulmonary lymphoid hyperplasia classically present in HIV-infected children older than 2 years of age. The imaging appearance of LIP includes nodules in a subpleural, septal centrilobular, or peribronchial distribution, and ground-glass opacities with or without lymphadenopathy [21] (Fig. 9). Other intrathoracic processes, such as thymic cysts and non-Hodgkin lymphoma, may occur in children with HIV infections.
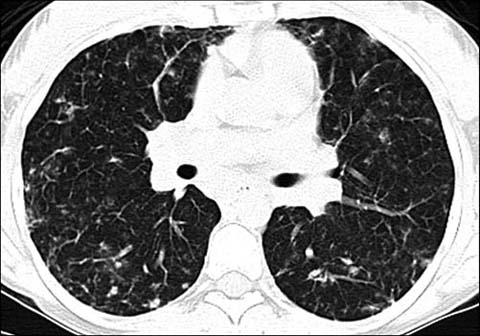
Fig 9.
An 18-year-old girl with lymphocytic interstitial pneumonitis. Axial lung window CT shows numerous small nodules and ground-glass opacities in both lungs
In patients with acquired neutropenia, the characteristic imaging findings of the “halo sign” and the “air-crescent sign” can help radiologists make an early and accurate diagnosis. The “halo sign” consists of a ground-glass halo of alveolar hemorrhage around a nodular or consolidative focus of infarcting lung from fungal vascular invasion and thrombosis; it strongly suggests invasive fungal disease (Fig. 10). An “air-crescent” sign presents as a crescent-shaped or circumferential area of radiolucency within a parenchymal consolidation or nodular opacity, and strongly suggests an underlying fungal ball (Fig. 11). It important to recognize that children with acquired immunodeficiency, particularly following allogeneic hematopoietic stem-cell transplantation, are susceptible not only to opportunistic pulmonary infections, but also to noninfectious pulmonary disorders including pulmonary edema, alveolar hemorrhage, drug reaction, bronchiolitis obliterans, graft-versus-host disease, and lymphoproliferative disease.
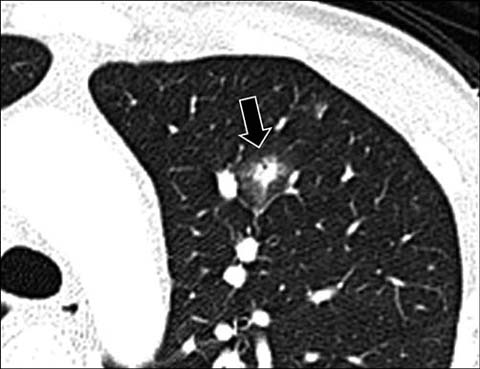
Fig 10.
A 14-year-old boy with leukemia. Axial lung window CT shows a round nodular opacity (arrow) surrounded by ground-glass opacity (“halo sign”) from invasive aspergillus infection
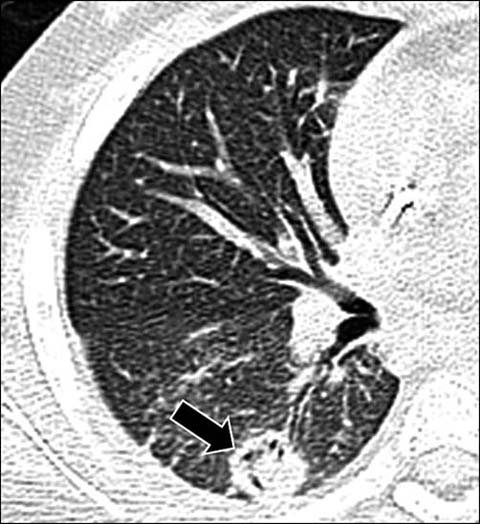
Fig 11.
A 10-year-old girl with aspergillus infection. Axial lung window CT shows a circumferential area of lucency (“air-crescent sign”) within a nodular opacity (arrow) due to an underlying fungal ball
Lysosomal Storage Diseases
Lysosomal storage diseases (LSDs) result from the absence or dysfunction of a lysosomal enzyme, with subsequent accumulation of the lipid glycol-protein, or mucopolysaccharide substrate. The three most commonly encountered LSDs in the pediatric population are Gaucher disease, Niemann-Pick disease, and mucopolysaccharidosis.
Gaucher disease, the most common LSD, is due to a congenital defect involving β-glucocerebrosidase. It results in the accumulation of glucocerebroside-laden macrophages in various organ systems, particularly the liver (hepatomegaly), spleen (splenomegaly and splenic infarcts), and bone marrow (osteonecrosis, endosteal scalloping, and Erlenmyer-flask deformities). Lung involvement is rare (∼2%) but, when present, small nodules, ground-glass opacities, and septal thickening can be seen [22]. Niemann-Pick disease is due to the deficiency of sphingomyelinase, which results in the accumulation of “foamy” macrophages. Lung involvement as seen on imaging studies is characterized by diffuse interstitial thickening due to lipid-laden macrophages infiltrating the pulmonary interstitium [23] (Fig. 12). A “crazy-paving” appearance, which refers to the appearance of ground-glass opacities with superimposed interlobular septal thickening and intralobular reticular thickening, can be seen on the CT scans of patients affected with the type C2 form. The mucopolysaccharidoses (MPS) result from the accumulation of glycosaminoglycans in multi-organ systems. Affected children typically present with upper- and lower-airway obstruction, restrictive pulmonary disease, and respiratory tract infections. Large-airway involvement in MPS reflects excessive glycosaminoglycan deposition in the airway walls, which may result in tracheobronchomalacia, airway-wall deformity, and airway luminal narrowing. Underlying concomitant skeletal dysplasia limiting chest wall motion and hepatosplenomegaly limiting diaphragmatic motion can cause extrinsic restrictive lung disease in affected children [24].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


