Cystic Fibrosis
Cystic fibrosis (CF) is a common inherited disease that has a high frequency in Caucasians. The disorder affects all exocrine glands, with symptoms involving the lungs and pancreas usually dominating the clinical picture. Even though the gene responsible for CF and its gene product, an integral membrane glycoprotein, have been identified, two aspects of the disease make CF particularly difficult to both diagnose and manage. First, there is tremendous variability in the degree and pattern of involvement of organs in different persons. In addition, we lack information about the precise details of the molecular and cellular pathogenesis of the disease. This chapter focuses on the pathophysiology and management of CF. Our current understanding of the genetics and underlying molecular biology is highlighted. Complications of the disorder are addressed, and a brief discussion of relevant psychosocial and reproductive issues is provided. Finally, potential future directions in treatment are described.
GENETICS
CF demonstrates an autosomal-recessive pattern of inheritance. In the United States, the incidence of the disease is approximately 1 in 3000 in Caucasians, 1 in 6000 in Hispanics, and 1 in 10,000 in African Americans. The frequency of unaffected heterozygote carriers of a CF mutation is estimated to be 1 in 26 in persons of Northern European ancestry.
CF is caused by mutations in a single gene named the cystic fibrosis transmembrane conductance regulator (CFTR). This gene was identified with an approach known as positional cloning, which permitted mapping of the gene, without prior knowledge of the biochemical defect, through use of polymorphic DNA markers. The first genetic marker that was found to be linked to CF was paraoxonase. In 1985, the demonstration of the linkage of CF to two DNA markers, D7S15 and D7S8, and to the met oncogene established the localization of the CF gene to the long arm of chromosome 7. Following a series of molecular cloning experiments, which included “chromosome walking” and “jumping,” a candidate gene was identified. This was proved to be the CF gene in 1989, largely through the discovery of a frequent mutation.1,2
The CF gene spans approximately 230 kb of DNA and contains 27 exons. The mRNA is 6.5 kb and is detected in a variety of tissues, including lungs, pancreas, and sweat glands, which are predominantly affected in pathogenesis of the disease. The deduced polypeptide was predicted to be an integral membrane glycoprotein containing 1480 amino acids (Fig. 50-1) (see “Pathogenesis” below). Several major and minor splicing variants in the transcripts have been described in individuals with and without CF. In most cases, however, the significance of the alternative splicings is not clear.
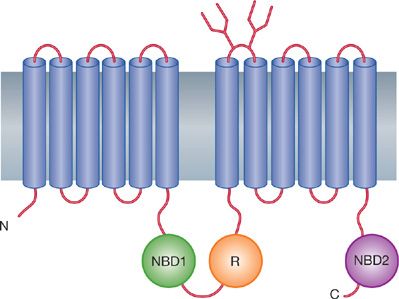
Figure 50-1 Domain model of the cystic fibrosis transmembrane conductance regulator (CFTR). Based on hydrophobicity plots, CFTR has 12 transmembrane-spanning domains, two nucleotide (N) binding domains (NBD 1 and NBD 2), and a regulatory (R) domain. The 12 transmembrane domains form the ion channel “pore.” In the closed state, the “R” domain is believed to obstruct the channel. Channel opening requires binding of two adenosine triphosphates (ATPs) to the nucleotide binding domains. This model is similar to other ATP-binding cassette transporter proteins that bind ATP and transport ions or micronutrients. (Modified with permission from Riordan J, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science; 245(4922):1066–1073.)
The most common CF mutation, and the first to be described, is a three-base deletion in exon 10 that causes a deletion of phenylalanine from position 508 (ΔF508 or F508del) of the CFTR glycoprotein. This mutation accounts for 66% of CF mutations.3 However, more than 1900 CF mutations have now been reported, and the list continues to grow. In addition, a number of benign sequence variations have been described. A listing of the most common mutations and their relative frequency is included in Table 50-1. The large number of mutations makes accurate detection of a satisfactory percentage of carriers extremely difficult, and carrier screening for the general population has not been recommended or implemented. Testing for 32 of the most common mutations is widely available; such testing will detect approximately 90% of the carriers in Caucasians of Northern European descent. In families with an affected individual and known mutations, prenatal diagnosis and carrier testing using direct detection of mutations is accurate and available. In families with a member diagnosed as having CF, but with undetected mutations, sequencing of the complete CFTR coding region and critical intronic regions is now also available to detect rare mutations.
PATHOGENESIS
Discovery of the gene responsible for CF and description of its product, CFTR, have provided the necessary foundation for understanding the pathogenesis of the disorder at the molecular and cellular levels. CFTR is an integral membrane glycoprotein of approximately 170 kD that is expressed in epithelial cells of affected organs. CFTR contains 1480 amino acids, which are arranged in 12 transmembrane domains, two nucleotide binding domains, and a putative regulatory domain (Fig. 50-1). The most common mutation, F508del, is a three-base deletion that causes deletion of phenylalanine from position 508, located in the proposed first nucleotide-binding domain. The original structural model, which was based on hydrophobicity plots, has proved to be essentially correct in its main features. CFTR shares many structural features with the “adenosine triphosphate (ATP)-binding cassette” transporter family, which includes P glycoproteins, as well as a number of bacterial transporters. CFTR has been clearly shown to function as an apical chloride channel in airway epithelial cells.
The localization of CFTR to the apical aspect of airway epithelial cells, to the ciliated duct of submucosal gland cells, and to submucosal serous cells, and the role of CFTR as an apical chloride channel fits nicely with the simplest hypothesis to account for the pathogenesis of pulmonary disease in CF. Decreased secretion of chloride and water by airway epithelial cells results in dehydrated mucus (Fig. 50-2). However, CFTR may have other functions, such as regulation of other ion channels, including the epithelial sodium channel. Loss of CFTR causes increased reabsorption of sodium; increased epithelium sodium channel activity alone alters regulation of ions and water, resulting in mucus obstruction of airways. CFTR transports bicarbonate; loss of CFTR function may result in acidification of the small intestinal lumen and, possibly, the airway lining fluid. Alternatively, CFTR may also function in intracellular membranes (e.g., endoplasmic reticulum, endosomes, phagosomes, and clathrin-coated vesicles). A consequence of the altered function of CFTR in intracellular membranes may explain abnormalities of CF glycoproteins: Increased sulfation of respiratory mucins, with decreased sialylation and increased fucosylation of both secreted and membrane glycoproteins. Altered glycosylation of airway glycoproteins may significantly impact bacterial–epithelial interactions and innate immune functions in the lung.4
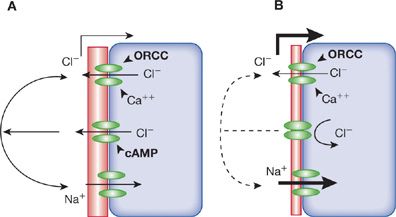
Figure 50-2 Simplified model of ion transport in airway epithelium. A. Normal airway cell with multiple apical ion channels. At the top, two different chloride channels are represented, the outwardly rectifying chloride channel (ORCC) and the Ca++-gated chloride channel. In the center, cyclic adenosine monophosphate (cAMP)-gated cystic fibrosis transmembrane conductance regulator (CFTR) is shown. The apical sodium channel is depicted at the bottom. Experimental data suggest that CFTR interacts with the other channels, although the type of interaction is not clear (solid arcs). B. CF cell with nonfunctioning cAMP-gated apical chloride transport. The function of the other channels is affected in an unknown manner (dashed arc). The net result of ion channel activity on the pericellular fluid composition (hatched area) is under investigation. Many questions remain concerning the function of CFTR and ion transport in the airway.
In addition to the effect of CFTR on epithelial ion channels and glycoprotein processing, loss of CFTR function negatively impacts innate immunity and accentuates inflammation. Absence of CFTR function is associated with impaired bacterial killing in vitro and defective function of antimicrobials including human β-defensin 1 and lysozyme. Absence of CFTR is also associated with increased interleukin-8 (IL-8) production and decreased IL-10 in vitro. In the CF airway, excessive neutrophil elastase cleaves complement and immunoglobulins, interfering with bacterial opsonization. CF airways have increased oxidant stress due to neutrophilic inflammation and reduced antioxidants such as glutathione. Together, these factors synergistically increase the inflammatory milieu in the airways in CF.
A long-standing impediment to progress has been the absence of a completely suitable animal model. A number of mouse models of CF have been developed and while these have been useful in understanding some features of the disease such as the regulation of inflammation, a drawback has been the absence of spontaneous development of lung disease.5 Two recently developed animal models in the ferret6 and pig7 have shown promise in developing some features of the early lung disease of CF.
CFTR mutations have been grouped into five classes,8 depending on the effect of the mutation on the expression, processing, and function of the protein (Fig. 50-3). The most common mutation, F508del, is a processing mutation in which very little of the mutant protein reaches the apical surface. If the mutant protein escapes normal intracellular processing, however, F508del protein functions normally in the apical membrane. Furthermore, only 25% of normal CFTR transcripts are properly processed and transported to the cell surface. The remaining 75% are degraded before being processed. These data suggest that one therapeutic strategy to overcome the defect in CF is to disrupt normal intracellular processing mechanisms. A phase III clinical trial is in progress to test a therapeutic small molecule which is based on this concept.
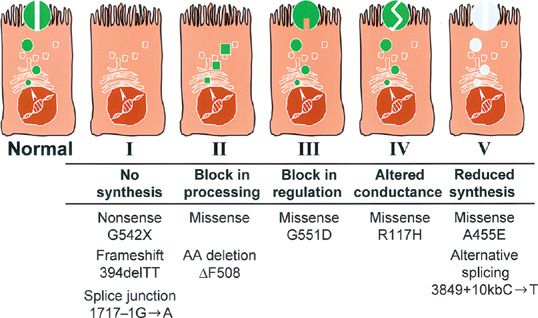
Figure 50-3 Classification of cystic fibrosis transmembrane conductance regulator (CFTR) mutations by molecular and biochemical abnormalities. This schematic depicts the effect of different classes of CFTR mutations on expression and function in the cell. Class I mutations block mRNA transcription. Class II mutations prevent normal CFTR protein processing and localization. Class III mutations permit CFTR localization at the apical membrane but inhibit chloride channel conductance. Class IV mutations result in partial chloride channel conductance. Class V mutations affect transcription, translation, or protein processing resulting in reduced CFTR expression at the apical membrane. Examples of mutations in each class are depicted below the cell models. Epithelial cell models with finger-like projections depict cilia at the apical surface. Fully processed CFTR protein is depicted by the gray circles embedded among the cilia at the apical surface of the cells. (Adapted with permission from Zielenski J1, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807.)
PATHOPHYSIOLOGY
In CF, all exocrine glands appear to be affected primarily, albeit to varying degrees. Because exocrine glands perform highly specialized functions in a variety of organs – for example, in the skin, respiratory tract, gastrointestinal tract, and reproductive system – the number of possible symptoms and complications in CF is large. Table 50-2 highlights the complications and symptoms of CF according to the age groups in which they most often occur.9 Obstruction of exocrine ducts by viscous secretions appears to play a cardinal role in the pathogenesis of almost all manifestations of the disease. In 10% to 20% of patients, the initial manifestation is often meconium ileus – that is, obstruction of the intestine by thick, viscous meconium stool. Chronic pulmonary disease, pancreatic insufficiency, and focal biliary cirrhosis progress gradually throughout the course of the disease, albeit at different rates in different patients. Progressive obstruction of exocrine ducts is a regular feature of the disease except in sweat glands, where obstruction of ducts has not been implicated in pathogenesis.
 RESPIRATORY TRACT
RESPIRATORY TRACT
In the lungs, hypersecretion of viscid mucus and chronic bacterial infection combine to produce a progressive and distinctive type of chronic obstructive airway disease that eventually leads to diffuse, severe bronchiectasis. The earliest pathologic lesions are found in the distal bronchioles. Whether the viscid secretions are primary or are secondary to chronic bacterial infections remain unsettled. In favor of a primary disturbance is the demonstration of mucus obstructing submucosal gland ducts in the airways of neonates with CF, who have not yet developed any evidence of bacterial infection or chronic colonization of the airways. With the use of sophisticated culture methods, bacterial pathogens can almost invariably be isolated from the respiratory tract of patients with CF. The most common pathogens isolated from sputum cultures are Staphylococcus aureus and Pseudomonas aeruginosa. Less commonly found are Escherichia coli, Klebsiella, and Haemophilus influenzae. In later stages of the disease, Pseudomonas usually predominates. By adulthood, more than 80% of patients are colonized with P. aeruginosa. Chronic infection with P. aeruginosa elicits an anaerobic milieu within mucus plugs in the CF airway.10 Using anaerobic culture conditions, large numbers of anaerobes, particularly Prevotella, Veillonella, and Propionibacterium, were detected in CF sputum but not in induced sputum from healthy volunteers.11 There is a correlation between P. aeruginosa–positive cultures and presence of the anaerobes. Multidrug-resistant organisms (MDROs) are detected in CF sputum cultures with higher prevalence and chronicity apparently associated with acute and chronic administration of antibiotics to suppress P. aeruginosa. These pathogens include Stenotrophomonas maltophilia, Achromobacter xylosoxidans, and Burkholderia cepacia complex. Other opportunistic pathogens including Aspergillus and nontuberculous mycobacteria have also been detected more commonly in CF sputum cultures. There is still controversy concerning whether these MDRO are actively contributing to CF lung disease or are commensal pathogens present in bronchiectatic airways.
Neutrophil-dominated lower airway inflammation also plays a primary role in the pathogenesis of the characteristic central bronchiectasis of CF.12 Bronchoalveolar lavage fluid (BALF) demonstrates increased neutrophils and various cytokines, especially IL-8, even in infants whose BALF is sterile.13,14
Typically, respiratory secretions increase when a patient with CF, already chronically colonized with Pseudomonas, develops a viral respiratory tract infection. In turn, the increase in secretions leads to a gradual increase in cough and sputum production and then to an exacerbation of the pulmonary disease, usually manifested by increase in respiratory rate; retraction of the chest during inspiration; and diffuse, coarse inspiratory crackles. Leukocytosis is common. The chest radiograph demonstrates worsening hyperinflation. Both peribronchial thickening and nodular or cystic densities are more marked than usual. Pulmonary function tests show a worsening over baseline. Usually, residual volume (RV) increases; forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) decrease; the forced expiratory flow between 25% and 75% of the exhaled vital capacity (FEF25–75%) also decreases. Treatment using antibiotics and chest physiotherapy generally succeed in restoring most indices of pulmonary function to, or almost to, baseline. However, Pseudomonas and Staphylococcus persist in sputum cultures.
The most attractive hypothesis to account for the pattern of response to treatment is that therapy reduces the number and, probably, virulence of organisms. Despite the virtual return to baseline after an exacerbation, however, the cumulative effect of repeated episodes is progressive bronchiectasis or atelectasis, or a combination of the two, accompanied by a gradual and irreversible decrease in pulmonary function. The striking degree of airway destruction and relative sparing of the pulmonary parenchyma at autopsy are shown in Figure 50-4. A simplified scheme illustrating the evolution of the process is shown in Figure 50-5.
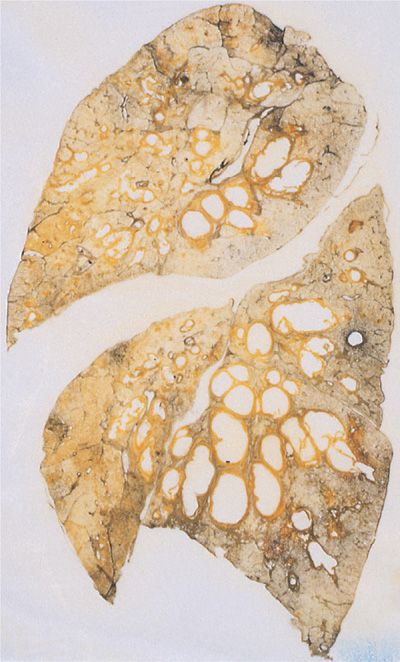
Figure 50-4 Section of lung from autopsy of a patient with CF, demonstrating remarkable dilation of large airways and preservation of intervening pulmonary parenchyma. (Used with permission of Dr. S. Moolten.)
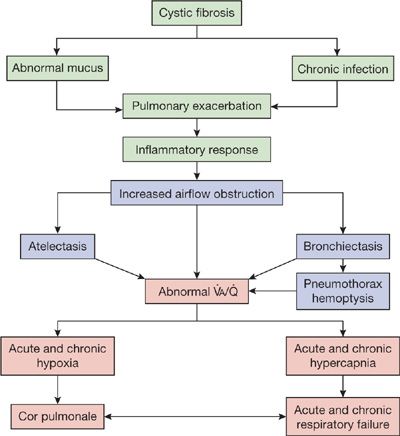
Figure 50-5 Simplified scheme for pathogenesis and progression of pulmonary disease in CF.
 GASTROINTESTINAL TRACT
GASTROINTESTINAL TRACT
Although pancreatic function may be either normal or abnormal at birth, it gradually becomes increasingly abnormal in most patients with CF as the pancreatic ducts become progressively obstructed by thick, viscous secretions from the exocrine portion of the organ; pancreatic enzymes that are trapped within the ducts lead to autodestruction of the pancreas. A cycle of destruction and obliteration of the ducts is set into motion, leading to cystic dilatation of ducts proximal to sites of obstruction and fibrosis of the body of the pancreas. In advanced stages of the disease, pancreatic fibrosis sometimes causes obliteration of the islets of Langerhans and, consequently, diabetes. This concept has been challenged by the notion that CFTR may have a direct effect on β-cell dysfunction as, discussed in the next section.
The liver and biliary tract are also affected in CF. Here too, the primary mechanism appears to be obstruction of small intrahepatic bile ducts by abnormally viscid secretions, leading to accumulation of toxic bile acids, depletion of hepatic antioxidants, and subsequent liver injury. CFTR is localized to the apical surface of the bile duct epithelium and not in the hepatocytes. Risk factors for liver disease appear to be male sex, meconium ileus, PiZ heterozygous state, and transforming growth factor-β1 (TGF-β1) polymorphisms. Elevated liver enzymes can be seen intermittently in 40% to 50% of patients with CF. Hepatic steatosis is frequently seen and may be related to malnutrition, essential fatty acid, choline and carnitine deficiency. Focal biliary cirrhosis, multilobular cirrhosis, and portal hypertension are also seen. Some newborn infants with CF develop the inspissated bile syndrome, characterized by prolonged obstructive jaundice starting at 2 to 8 weeks of age. The jaundice often clears without therapy. Gall bladder anomalies may be seen in about 24% to 50% of patients and include microgall bladder, gall stones, and gall bladder dysfunction.15 The most striking pathologic change in the intestines is hyperplasia of the mucus glands and goblet cells. Biochemical abnormalities in intestinal mucins may contribute to malabsorption of specific nutrients and bile acids. Much of the malabsorption in CF can be corrected by administration of pancreatic enzyme replacement therapy (PERT). However, the abnormal mucins may lead to slowing of intestinal transit time; the slowing, combined with maldigestion of food substances, sometimes causes fecal impaction in the terminal ileum and ileocecal area, a condition referred to as meconium ileus equivalent or distal intestinal obstruction syndrome. The fecal impaction, in turn, occasionally causes volvulus or intussusception of the bowel (Fig. 50-6).
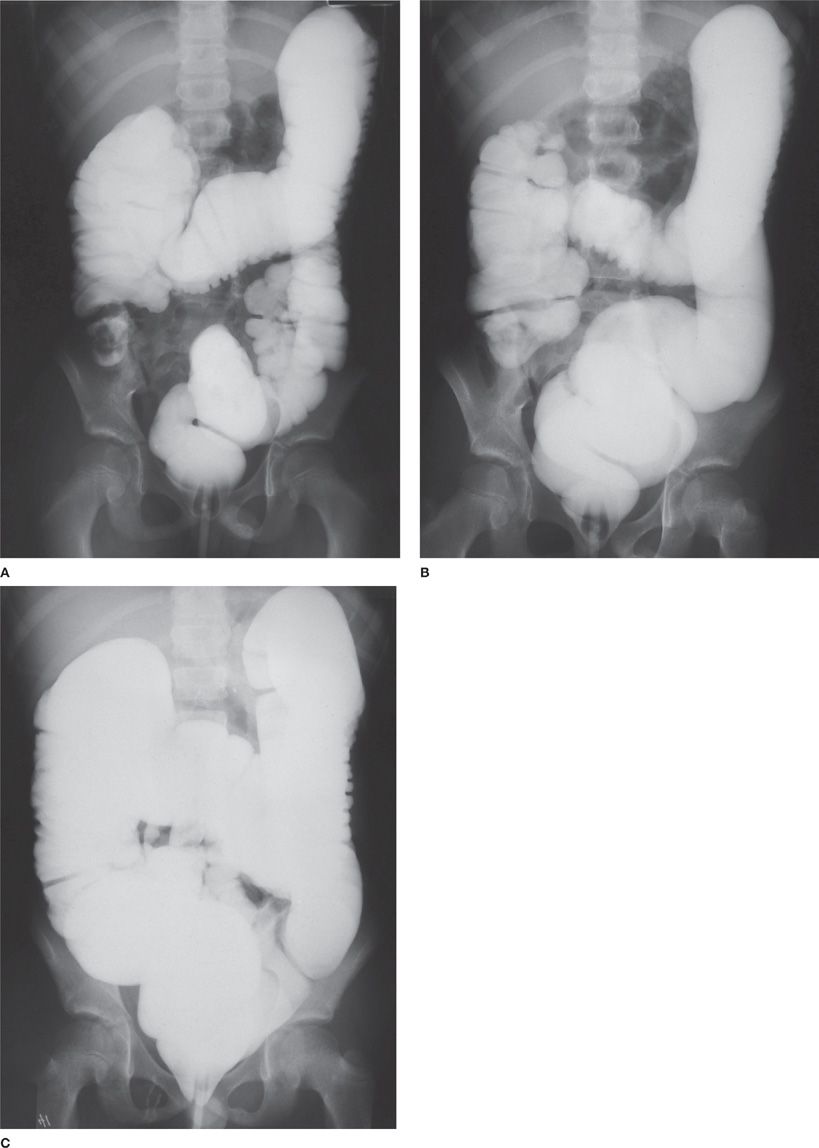
Figure 50-6 Distal intestinal obstruction syndrome (DIOS). A. Presenting Gastrografin enema of a child who had crampy abdominal pain and a right lower–quadrant mass. Fecal impaction with intussusception is demonstrated. B. Partial resolution of the obstruction following Gastrografin administration. C. Complete resolution of the intussusception and fecal impaction.
 ENDOCRINE INSUFFICIENCIES
ENDOCRINE INSUFFICIENCIES
Important CF-associated endocrine disorders are discussed below.
Cystic Fibrosis–Related Diabetes
Diabetes mellitus is a common comorbidity in CF and increases in frequency with increasing age. According to data from University of Minnesota where annual cystic fibrosis–related diabetes (CFRD) screening is recommended for all patients ≥6 years, CFRD affects 2% of children, 19% of adolescents, and 40% to 50% of adults.16 Similarly, CFRD was found in over a third of 775 patients of age ≥6 years undergoing CFRD screening over a 15-year period.17
Underscoring its relevance, CFRD has been associated with decreased survival,18–24 worse pulmonary function, and lower BMI.23 Declines in pulmonary function and nutritional status have been observed even prior to the CFRD onset.22,25 Moreover, less significant glucose impairments are associated with greater declines in nutritional status over the previous year in children25; these findings suggest more subtle glucose abnormalities may be clinically relevant in CF. Microvascular complications, such as retinopathy, nephropathy, and neuropathy also occur in CFRD but may be limited to those individuals with fasting hyperglycemia (FH).17
Fortunately, early identification and treatment of CFRD appear to curb the impact of CFRD upon survival.16 Insulin treatment of adults with CFRD without FH improves BMI,26 and small studies have found insulin treatment improves weight in children even prior to development of CFRD.27–29
As described above, CFRD is considered an insulin-deficient state. In fact, delayed and blunted insulin secretion in response to a glucose load or meal prevails even in the setting of “normal” glucose tolerance in CF,30–32 and these abnormalities progress with worsening glucose intolerance.30,32,33 This progressive decline in insulin secretion has traditionally been considered a product of “collateral damage” extending from obstructive damage to the exocrine pancreas.
This “bystander” model has been challenged in more recent years. Animal models suggest CFTR may play a direct role in β-cell dysfunction.34,35 For instance, CFTR knockout ferrets demonstrate glucose abnormalities and insulin secretion defects as newborns.35 Moreover, the T2DM GWAS-implicated gene, TCF7L2, which may contribute to the defective insulin secretion that underlies T2DM, confers an even stronger risk of CFRD. In addition, disturbances in secretion/function of incretins, gut-secreted hormones that potentiate insulin secretion, have been described in CF36 and in T2DM.37–40,44–47
Osteoporosis/Vitamin D Deficiency
Low bone mineral density occurs in as many as 85% of adults with CF. The origin of osteoporosis in CF is multifactorial and is attributed to pancreatic insufficiency and malnutrition and poor growth, CFRD, deficiencies in vitamin D, vitamin K, and calcium levels, elevated inflammatory cytokines, pubertal delay, diabetes, and exposure to glucocorticoids.41 Bone histology in clinically stable CF adults is significant for decreased cancellous bone volume and decreased connectivity.42 Importantly, the sequelae of low bone density in CF patients include increased risk of vertebral and rib fractures, approximately twice as great as the general population, and increased risk of kyphosis.
 REPRODUCTIVE ORGANS
REPRODUCTIVE ORGANS
Except for an increase in viscosity and an abnormal midcycle ferning pattern in cervical mucus, no consistent pathologic changes occur in the female reproductive tract in patients with CF. In the male reproductive tract, however, the vas deferens is either atretic or absent at birth. Although the pathogenesis of this lesion is not certain, viscous secretions may contribute to obstruction in utero, followed by failure of development of the vas deferens. Spermatogenesis and testicular development are otherwise normal. Because of either partial or complete obstruction of the vas deferens, approximately 98% of males with CF are aspermic.
 SWEAT GLANDS
SWEAT GLANDS
The sweat glands of patients with CF manifest no distinctive histologic changes. Nonetheless, their function is abnormal. Micropuncture experiments have shown that the precursor solution secreted by the sweat glands is isotonic to plasma, both in CF patients and in normal subjects. In normal persons, as the sweat flows along the duct of the gland, sodium and chloride are reabsorbed, so that by the time the opening at the skin surface is reached, sweat is hypotonic to plasma with respect to both sodium and chloride concentrations. In CF, the relative impermeability to chloride ions is thought to be responsible for the elevated chloride and sodium concentrations which are the basis for the diagnostic test, the quantitative pilocarpine iontophoresis sweat test, and are also responsible for the characteristic increase in potential differences across isolated, perfused sweat glands from CF patients.
DIAGNOSIS
The diagnosis of CF requires the demonstration of abnormally high concentrations of sodium and chloride in the sweat of a person who has the characteristic history and symptoms of CF. The most prominent clinical features are chronic pulmonary disease and pancreatic insufficiency. The most compelling family history for the diagnosis is CF in a sibling. If the clinical picture and/or the family history support the diagnosis, and if two sweat tests using the quantitative pilocarpine iontophoresis method are clearly positive, the diagnosis of CF can be made with assurance. Identification of two pathologic mutations, in addition to the characteristic clinical picture, is accepted as a criterion for the diagnosis. However, CF is a complex syndrome (Table 50-2) whose clinical manifestations are sometimes subtle. In addition, the family history is not always straightforward. Therefore, a high index of suspicion, coupled with a battery of clinical tests, is sometimes required to establish the diagnosis, especially in adolescents or young adults.
Since CF occurs with a high frequency in the general population, the diagnosis should be considered routinely in a broad array of differential diagnoses. Although Table 50-2 categorizes symptoms according to the age at which they most often occur, symptoms at any age should prompt consideration of the diagnosis of CF.
The most consistent feature of CF is an abnormally high concentration of sodium and chloride in sweat. Measurement of the chloride concentration is recommended for clinical testing. The only reliable sweat test is based on iontophoresis of pilocarpine, followed by quantitative determination of the concentration of chloride in an adequate, measured volume of sweat. Guidelines for the proper performance of a sweat test have been published.43 In children, concentrations of chloride of less than 40 mEq/L are usually regarded as normal. However, the average of values for sodium and chloride concentrations is about 20 mEq/L for normal subjects and 95 mEq/L for those with CF. In children, values between 40 and 60 mEq/L are traditionally considered borderline elevated; such values call for further evaluation. As a result of recent experience with CF newborn screening, it has been suggested that sweat chloride values above 30 mEq/L may be diagnostic in the first few months of life.
The concentration of sodium and chloride in sweat increases gradually with age. Conditions other than CF in which the concentrations of sodium and chloride in sweat are abnormally high include malnutrition, adrenal insufficiency, hereditary nephrogenic diabetes insipidus, ectodermal dysplasia, and fucosidosis. Except in some instances of malnutrition, these conditions are readily distinguished from CF.44 The finding of abnormal concentrations of sodium and chloride in sweat should automatically prompt evaluation of the patient to determine if, and to what extent, other organs are affected.
Genetic analysis can be used to confirm the diagnosis of CF. In patients with minimal symptoms, the diagnosis of CF can be made with certainty if two CF-associated alleles are present. As mentioned previously, screening for 32 of the most common alleles yields an overall sensitivity of 90% due to undetected alleles. Therefore, a negative mutation analysis does not rule out a diagnosis of CF, and atypical patients should be followed carefully.
Newborn screening is now standard practice in the United States.45 The initial stage of screening often uses the neonatal blood spot to determine the concentration of immunoreactive trypsinogen. If this is elevated, secondary screens vary in individual states from repeat immunotrypsinogen determination to F508del or 25 to 32 mutation screen. The screening programs have a sensitivity ranging from 87% to 99%. Risks versus benefits and the relative costs of the screening programs are being evaluated to determine the best approach. Infants with positive newborn screens for CF are referred to CF centers for sweat test confirmation. It has been proposed that infants who have a positive newborn screen, but do not otherwise fulfill the criteria for a diagnosis of CF,45 should be termed as having “CFTR metabolic syndrome”46,53 and at a minimum they should be followed in a CF center until their status can be clarified.
CLINICAL EVALUATION
The evaluation of patients with CF includes chest radiography, tests of pulmonary performance, sputum culture, and assessment of pancreatic, endocrine, hepatic, and reproductive functions. Each is described below.
 PULMONARY ASSESSMENT
PULMONARY ASSESSMENT
Pulmonary assessment includes chest radiography; measurement of pulmonary function, including that of small airways; and evaluation of gas exchange.
Chest Radiography
Rarely is the chest radiograph completely normal in CF. In the person with minor pulmonary symptoms, the manifestations may be questionable (e.g., mild hyperinflation and minimal peribronchial thickening). However, the radiographic findings become more distinctly abnormal as the disease increases in severity. Peribronchial thickening, which is often most prominent in the upper lobes of the lungs early in the course of the disease, usually progresses to affect all lobes. In the advanced stage of pulmonary involvement, ring shadows, cystic lesions, and nodular densities are increasingly apparent, as are areas of bronchiectasis and atelectasis. The central pulmonary artery often enlarges in the middle stages of the disease, but the cardiac silhouette remains within normal limits until the disease is far advanced. The variability in the chest radiograph is illustrated in Figure 50-7 for three siblings with CF when each was 17 years old.
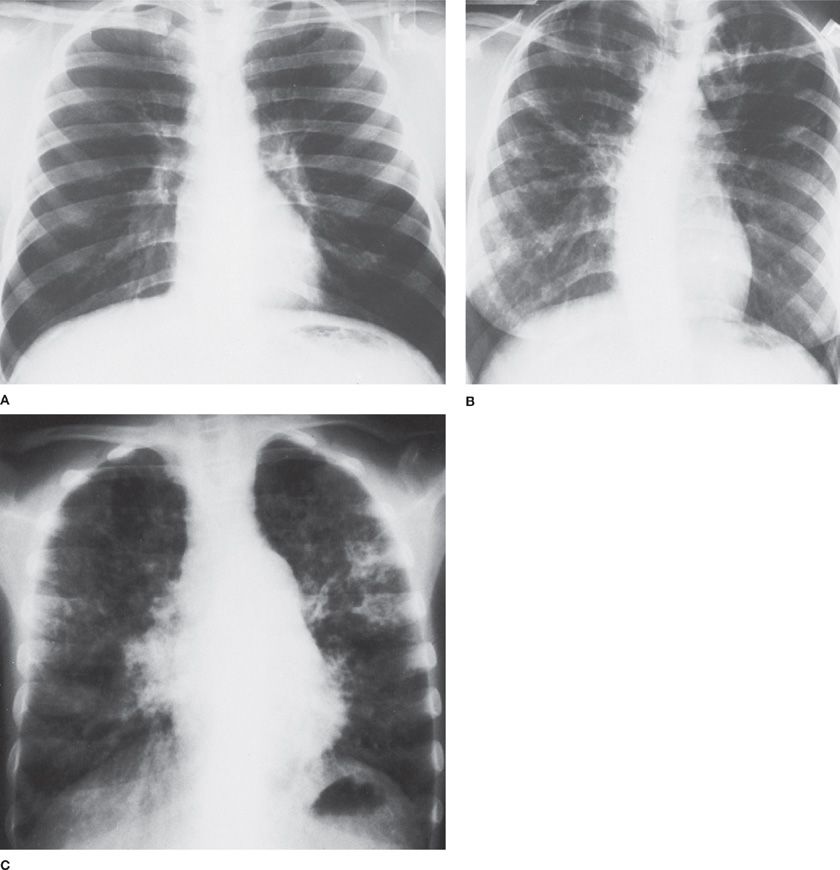
Figure 50-7 Chest radiographs of three siblings with CF taken when the patients were 17 years of age. A. Mild hyperinflation; otherwise normal. Patient is now 32 years old and has been hospitalized once for treatment of electrolyte depletion. B. Diffuse peribronchial thickening, mild hyperinflation, and cystic changes in both upper lobes. The patient was hospitalized seven times for pulmonary exacerbations, once for diabetes, and once for hemoptysis. She died at age 34 following complications from lung transplantation. C. Severe hyperinflation, diffuse peribronchial thickening, multiple infiltrates, and increased pulmonary vascular markings and heart size. The patient died 1 month later from respiratory failure complicated by congestive heart failure.
High-resolution computed tomography (HRCT) scans are more sensitive than plain radiographs. The most common abnormalities described are bronchiectasis, peribronchial thickening, mosaic perfusion, air trapping, and mucus plugging.47 Early bronchiectasis is easily detected on the CT scan, even when routine chest radiographs are normal, as seen in Figure 50-8. CT scan abnormalities may be detected prior to a change in pulmonary function tests. Chest CT scan abnormalities have been detected in asymptomatic newborn infants diagnosed by newborn screening.48 Currently, there are no standard recommendations for CT use due to the radiation exposure incurred by these studies.
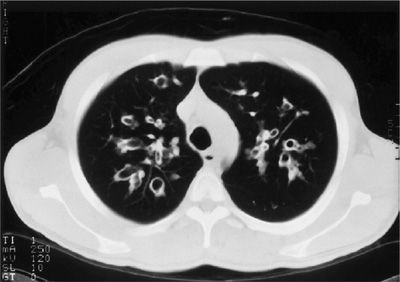
Figure 50-8 High-resolution chest computed tomography scan from a patient with CF. Marked bronchiectasis with peribronchial thickening is shown in the upper lobes.
Pulmonary Performance
The lungs of patients with CF are usually morphologically and functionally normal at birth. Over time, accumulation of tracheobronchial secretions and recurrent infections progressively impair pulmonary function in almost all patients. In the fully developed clinical syndrome, all the pulmonary function abnormalities seen in chronic bronchitis, emphysema, and asthma may occur. However, one complicating regular feature of CF – bronchiectasis – modifies pulmonary performance. Chronic, local infection and airway damage increase the compliance of bronchiectatic airways, resulting in airway collapse during rapid expirations or cough. The usefulness of pulmonary function testing in CF is twofold: Tracing the natural history of the disease and assessing the value of therapeutic interventions.
The earliest stages of the pulmonary disorder are the most difficult to quantify. In infants, tests are limited almost entirely to those that do not depend on the patient’s understanding and cooperation. A variety of methods to measure infant pulmonary function have been devised; one method, the raised volume rapid thoracoabdominal compression technique, requires sedation of infants but provides values most similar to standard spirometric values, and has detected reduced pulmonary function in infants with CF49 Nitrogen multiple breath washout or lung clearance index, can be performed in preschool age children and detects early ventilation inhomogeneity that correlates with airflow obstruction by spirometry.50 After age 6, pulmonary function tests originally designed for adults may be performed quite readily on children. Changes in pulmonary performance throughout the natural history of CF can be described with confidence.
Obstruction in Small Airways
The small airways – that is, the bronchioles – are vulnerable to obstruction early in the course of CF. At this stage, as in cigarette smokers, results of tests for small airway disease are apt to be abnormal, while those of tests for obstruction of large airways are still normal. Three factors interact in causing the obstruction: (1) Intrinsic disease of the smaller airways, often in association with bronchiectasis in the proximal, larger airways; (2) viscid secretions, impaired ciliary action, and impaired cough; and (3) progressive decrease in lung elastic recoil.
The progressive reduction in lung elastic recoil in CF is predominantly a function of overinflation due to intrinsic airway disease, rather than loss of pulmonary parenchyma. This mechanism differs from that in chronic bronchitis and emphysema, in which the combined effects of parenchymal destruction and overinflation are responsible for the decrease in elastic recoil. Emphysema is not a regular feature of CF. In some patients, emphysema occurs only late in the course of the disease (Fig. 50-4).
Airway smooth muscle tone increases only slightly in CF. Exercise elicits bronchodilation, followed shortly thereafter by bronchoconstriction. Both the bronchodilation and bronchoconstriction are far less impressive in CF than in asthma. Indeed, exaggerated bronchomotor responses in CF raise the possibility of superimposed asthma. In distinguishing between contributions to airway obstruction by intrinsic airway disease caused by CF and asthma, maximal expiratory flow–volume curves are sometimes helpful.
Because of the bronchiolar locus of the early lesions in CF, abnormalities in breathing frequency–dependent tests (e.g., dynamic lung compliance), in volume-dependent tests (e.g., closing volume), and in maximal expiratory flow (![]() Emax) at low lung volumes are demonstrable, even though results of tests of large airway function (e.g., FEV1 and airway resistance) are still normal.
Emax) at low lung volumes are demonstrable, even though results of tests of large airway function (e.g., FEV1 and airway resistance) are still normal.
Change in Lung Volumes
As with chronic bronchitis, emphysema, and asthma, RV in CF increases. Thereafter, an increase in functional residual capacity and, sometimes, in total lung capacity is seen. As CF lung disease progresses, air trapping increases in severity and is manifest as an elevated ratio of RV to total lung capacity. This change decreases the compliance of the lung and increases the work of breathing.
Abnormalities in Gas Exchange
Early in the evolution of the pulmonary abnormalities in CF – that is, when tests of small airway function alone are abnormal – ventilation–perfusion abnormalities usually result in widening of the alveolar–arterial oxygen gradient and an increase in the ratio of dead space to tidal volume (VD/VT). These abnormalities portend increasing inhomogeneities in alveolar ventilation and blood flow as the affected child grows to adulthood. The diffusing capacity for carbon monoxide (DLCO) is low at rest and does not increase normally during exercise. This observation is difficult to reconcile with the preservation of the gas-exchanging surface of the lungs (in the absence of emphysema) until late in the course of the disease (Fig. 50-4).
As obstructive disease of the airways progresses and exaggerates the imbalances between alveolar ventilation and blood flow, arterial hypoxemia develops; pulmonary hypertension, cor pulmonale, and right ventricular failure follow, in turn. Late in the course of the disease, hypercapnia and respiratory acidosis contribute to the final picture of respiratory failure. At this juncture, the ventilatory response to inhaled CO2 is depressed. Bouts of infection punctuate the course of the illness; during each episode, pulmonary function deteriorates, but it usually returns toward baseline, except in the preterminal stages of the disorder.
 SPUTUM CULTURE
SPUTUM CULTURE
The unique respiratory flora isolated from sputum or oropharyngeal cultures from patients with CF is helpful in establishing the diagnosis and in guiding the antimicrobial therapy for acute exacerbations. In many patients with CF, P. aeruginosa and S. aureus are found alone, or in combination with other organisms, in the sputum. There is growing evidence of correlations between different organisms and CF lung disease progression. The presence of mucoid Pseudomonas is important because acquisition of mucoid Pseudomonas predicts more rapid progression of CF lung disease.51 Similarly, acquisition of methicillin-resistant S. aureus is associated with a decline in pulmonary function52 and worse survival.53 Infection with B. cepacia complex organisms may be aggressive with rapid deterioration of clinical status or may have an indolent course; the presence of mucoid-positive organisms appears to be protective.54 Chronic antibiotic therapy to suppress Pseudomonas has improved clinical outcomes but has also led to a greater number of MDROs identified in sputum, including S. maltophilia and A. xylosoxidans. Chronic colonization with S. maltophilia, induces a serologic response and independently correlates with progression in airflow obstruction.55
In addition to bacteria, sputum cultures for detection of molds and nontuberculous mycobacteria are helpful to guide therapy for patients with acute exacerbations unresponsive to antibiotic therapy, or for patients with unexplained progression of lung disease. Allergic bronchopulmonary aspergillosis (ABPA) complicates CF lung disease,56 and recent evidence suggests that Aspergillus fumigatus may also be associated with airway infection or allergy-triggered asthma in the absence of ABPA.57 The prevalence of nontuberculous mycobacteria ranges from 7% to 24% with the most frequent species identified as Mycobacterium avium complex and Mycobacterium abscessus.58
 PANCREATIC FUNCTION
PANCREATIC FUNCTION
The evaluation of pancreatic function is an important part of establishing the diagnosis of CF, since almost 90% of patients have pancreatic insufficiency. Infants with pancreatic insufficiency due to CF can present with failure to thrive and loose or frequent stools. However, visual appearance does not always correlate with the degree or presence of fat malabsorption. Currently the diagnosis of pancreatic insufficiency can be made by measuring fecal elastase (FE)-1 levels and assessment of the degree of malabsorption is best accomplished by the determination of the coefficient of fat malabsorption. The FE-1 test is performed on a random single stool sample. It is easy to obtain and in patients with CF, a human enzyme-linked immunosorbent assay (ELISA) for FE has a sensitivity of 98% to 100% and a specificity of 93% to 100%, even while patients are taking pancreatic enzyme supplements.59 Also available is a polyclonal assay that detects human as well as porcine elastase and should not be used to diagnose pancreatic insufficiency in patients already on pancreatic enzyme supplementation. There is some debate as to the value used for the diagnosis of pancreatic insufficiency.60 The coefficient of fat absorption is performed by collecting stools for 72 hours while the patient is ingesting a high-fat diet (documented on a 3-day diet record) and analyzing the stool fat content. A malabsorption coefficient of greater than 7% is usually considered abnormal. Patients with CF usually have a malabsorption coefficient around 20% to 30%. The test is not popular with families who find the stool collections very unappealing. Pancreatic stimulation tests are the most accurate measurement of pancreatic function but are invasive, cumbersome, and at this time are not clinically available.
Cystic Fibrosis–Related Diabetes
Because the onset of CFRD is generally insidious, and FH tends to be a late manifestation, annual CFRD screening with an oral glucose tolerance test (OGTT) starting by age 10 is recommended.61,62 Based upon the fasting, 1-hour, and 2-hour plasma glucose (PG0, PG1, and PG2) during the OGTT, the following glucose tolerance categories are defined.
• Normal glucose tolerance (NGT) = PG1 <200 mg/dL and PG2 <140 mg/dL
• Indeterminate = PG1 ≥200 mg/dL but PG2 <140 mg/dL
• Impaired glucose tolerance (IGT) = PG2 ≥140 and <200 mg/dL
• CFRD = PG2 ≥200 mg/dL
Without FH = PG0 <126 mg/dL
With FH = PG0 ≥126 mg/dL
Contrary to what one might expect, isolated impaired fasting glucose (PG0 100–125 mg/dL) is not associated with worse survival, nutritional status, pulmonary function, or progression to CFRD.63 Over 10 years, FH occurs in 60% of patients with CFRD without FH at baseline.17 At least in children, increased plasma glucose at 1 hour during the OGTT predicts increased risk of progression to CFRD.64
Additional CFRD screening measures include fasting and postprandial glucose measurements during hospitalizations for acute illness.61,62 Home glucose monitoring should also occur periodically during and after continuous overnight enteral feeds as well as during intercurrent illnesses, intravenous antibiotic therapy, and glucocorticoid treatment.65 While an elevated hemoglobin A1C (>6.5%) is consistent with CFRD, the HbA1C tends to underestimate overall glucose intolerance in CF patients and is not generally recommended for CFRD screening.
Osteoporosis/Vitamin D Deficiency
Patients with CF are at risk for vitamin D deficiency and osteoporosis. Annual serum 25-hydroxy vitamin D levels should be monitored. In addition, in all adults and children greater than 8 years with risk factors for osteopenia including malnutrition, chronic glucocorticoid use, moderate-to-severe airway obstruction (FEV1 <50% predicted), or history of fracture or delayed puberty, a DXA scan should be obtained to monitor bone mineral density.41
 LIVER FUNCTION
LIVER FUNCTION
Evaluation of liver function (transaminases, bilirubin, gamma glutamyl transferase [GGT]) is an important part of the evaluation of CF. Transient elevations of serum transaminases can be often seen and may be related to intercurrent illnesses and medications. However, these tests can often be relatively normal, even in patients with mild or moderate focal biliary cirrhosis. The prothrombin time is sometimes prolonged, owing to a combination of malabsorption and decreased synthesis of clotting factors by the liver. Obtaining a level of protein induced by vitamin K absence-II (PIVKA-II) to assess vitamin K status can be helpful in these instances. A liver ultrasound with Doppler should be performed in patients with persistently elevated liver function tests. Fatty liver, cirrhosis, splenomegaly, varices, and reversal of portal blood flow can be seen on ultrasound. Occasionally, patients present with bleeding esophageal varices from advanced cirrhosis and an upper endoscopy is helpful diagnostically and therapeutically.
 SEMEN ANALYSIS
SEMEN ANALYSIS
Occasionally, a man who is found to have aspermia during the course of an evaluation for infertility is found to have CF. In men with CF, a complete semen analysis is part of the evaluation. Azoospermia is found in more than 98% of men with the disorder.
 MUTATION ANALYSIS
MUTATION ANALYSIS
There are currently over 1900 mutations associated with CF (http://www.genet.sickkids.on.ca/cftr/). A new initiative, The Clinical and Functional TRanslation of CFTR (CFTR2) (www.cftr2.org), funded by NIH, the US CFF, and Sequenom, is a website dedicated to publishing the functional implications of CFTR mutations. Patients homozygous for the most common mutation, F508del have pancreatic insufficiency; patients with CF who have pancreatic insufficiency tend to have a worse prognosis. F508del is one of the major mutations classified as disease causing; other mutations are associated with CF-related disorders, and yet other mutations have no known clinical importance or unknown significance. Several mutations, including R117H, are associated with pancreatic sufficiency and a mild phenotype. Interestingly, the phenotype of R117H is linked to the expression of the polyT and polyTG intronic domains found 5’ to exon 9. A T5 polymorphism expressed with R117H results in congenital bilateral absence of the vas deferens (CBAVD) or idiopathic pancreatitis, and may be complicated by mild lung disease. In contrast, R117H associated with T7 or T9 may have no manifestations of CF or CF-related disease.66
Certain alleles associated with CF (e.g., 3849+10kbC→T) are associated with nasal polyposis and bronchiectasis but normal sweat test results. The diagnosis of CF can be made with confidence in these patients. More problematic are persons with atypical presentations, normal sweat test results, and at least one CF-associated mutation. More extensive genotyping should be attempted for all patients with a high clinical suspicion for CF (see “Genetics”) because mutation analyses will be used to determine eligibility for mutation-specific protein-correcting therapies. For example, therapy for CF patients with the G551D mutation with the CFTR potentiator ivacaftor® results in astonishing improvements in respiratory tract symptoms, weight gain, and shift from positive to borderline sweat chloride values (see “Therapy,” “Genetics,” and “Future Directions”).67
Patients with the same genotype may have dramatically different phenotypes, supporting the concept that modifier genes play an important role in determining the CF phenotype. Three large GWAS consortia are collaborating to define modifier genes associated with lung disease severity68 and with specific disease manifestations such as CFRD,69 liver disease,70 and meconium ileus.71 Investigations using these large well-characterized cohorts recruited by the consortia have identified chromosomal regions of interest but have not yet confirmed gene targets or polymorphisms that confer increased risk of severe lung disease. Using a hypothesis-driven approach, several potential candidate modifier genes for severity of lung disease were identified, including α1-antitrypsin, HLA antigens, nitric oxide synthase, mannose-binding lectin, TGF-β, tumor necrosis factor-α (TNF-α), and β2-adrenergic receptor. However, none of these genetic polymorphisms were validated as gene modifiers by GWAS analysis in the combined consortia.
ATYPICAL CLINICAL PRESENTATIONS
Atypical clinical presentations confound the diagnosis of CF in adults; a high index of suspicion is required to establish the diagnosis. Approximately 6% of all CF is diagnosed after age 18. Late presentations of CF tend to occur in persons with pancreatic sufficiency; indeed, overweight or well-nourished persons may have CF. Recovery of unusual gram-negative organisms, mucoid Pseudomonas species, or S. aureus from sputum of asthmatics with persistent sputum production, chest radiographic abnormalities, or clubbing should prompt referral for sweat testing. Recurrent sinusitis and nasal polyposis may be the only manifestations of CF in a mildly affected person. Isolation of P. aeruginosa from deep nasal cultures should raise the suspicion of CF. Frequently, the sinus findings on CT mimic fungal sinusitis, demonstrating concentric, inhomogeneous material. Occasionally, persistent inflammation produces bony destruction that is mistaken for previous surgical intervention. Sweat testing and referral to a CF center should be considered for men with azoospermia or CBAVD. The clinical entity, “CFTR-related disease” has recently been defined by an international working group. CFTR-related disease encompasses patients with evidence of CFTR dysfunction who do not meet the criteria for CF disease (e.g., only one CFTR mutation, a normal sweat test, and single organ involvement). CFTR-related diseases include CBAVD, idiopathic pancreatitis, and diffuse bronchiectasis.72
TREATMENT
Intensive, comprehensive CF treatment programs designed to deal with particular symptoms, correct deficiencies, and prevent progression and complications of the disease have led to a dramatic increase in the median age of survival. Although the value of comprehensive treatment is beyond question, far less certain are the utility of each component of the treatment plan and the level of each component necessary in a given patient. At present, the best approach still appears to be determination of the type and degree of abnormality in individual patients and design of a treatment program that will improve or maintain function of the organ systems affected. Two recent reviews highlight the evidence supporting each component of CF therapy.73,74 To ensure that the treatment regimen meets the needs of the individual patient, that necessary treatment is not omitted, or that side effects of prescribed treatments do not go unnoticed, it is often desirable to hospitalize the patient for diagnosis and evaluation. Hospitalization also provides an excellent opportunity for counseling the patient, parents, and family about the diverse aspects of the diagnosis, treatment, prognosis, and inheritance pattern of CF. Hospitalization provides the opportunity to monitor the response of individual patients to each component of the therapeutic program.
An important aspect of the care of patients with CF is the network of more than 100 CF centers that exist throughout the United States and the larger network throughout the world. CF centers use a team approach to the care of patients. A CF care team usually includes physicians, nurses, respiratory therapists, physical therapists, nutritionists, social workers, and genetic counselors.
 MANAGEMENT OF PULMONARY DISEASE
MANAGEMENT OF PULMONARY DISEASE
Management of CF-related pulmonary disease focuses on chronic respiratory management and treatment of acute exacerbations.
Chronic Maintenance Therapy
More than 90% of the patients with CF die from respiratory failure or pulmonary complications. The goals of treating the pulmonary disorder in CF are to prevent and treat the complications of airway obstruction and infection. Although management of the pulmonary disorder consists of many components applied in combination,75 the individual components of therapy are discussed separately below.
Chest Physiotherapy
Almost all treatment programs for CF include a strategy intended to clear pulmonary secretions to prevent complications arising from airway plugging by viscous secretions. Chest physiotherapy – that is, “percussion and postural drainage” – performed regularly, is the most widely prescribed method. In infants and young children, chest physiotherapy is generally performed routinely, twice daily. Percussion and postural drainage technique has been modified to exclude head-down positioning which increased the risk of GE reflux and increased the duration of acute exacerbations of cough in infants.76 In addition to manual chest percussion and postural drainage, there are several other effective modalities for chest physiotherapy. These alternative measures include the high-frequency chest-wall oscillation (HFCWO) vest; the flutter device, a small pipe–like device that produces an oscillating resistance during a forced expiratory maneuver; the acapella device that produces both positive expiratory pressure (PEP) and an oscillating resistance during forced expiratory maneuver; PEP mask; intrapulmonary percussive ventilation; autogenic drainage and active cycle of breathing; and exercise.77
Overall, due to the difficulty in designing unbiased, well-controlled trials with sufficient power to unequivocally distinguish efficacy between treatments, evidence does not exist to demonstrate superiority of one airway clearance mechanism over another. Therefore, the recommendations based on expert opinion are that airway clearance should be initiated in asymptomatic infants because they develop signs of lung disease within the first few months of life, and that patients should be offered and instructed in a variety of methods so that they can select the methods that they deem provides the most subjective benefit.78 Some form of physiotherapy that is effective in mucus clearance is required daily because without chest physiotherapy, pulmonary function deteriorates.79 At present, most CF centers recommend that all patients with CF attempt to maintain clearance of pulmonary secretions with a method that is applied regularly (e.g., twice daily). An additional recommendation is that chest physiotherapy be applied more often during an exacerbation of the chronic pulmonary infection. Unfortunately, the recommendation of chest physiotherapy on a regular basis – a time-consuming and often arduous form of treatment – is difficult to implement without considerable support and encouragement from family and health professionals.
Mucolytics and Inhaled Hypertonic Saline
A number of mucolytic agents have been tried over the years. One that has endured is N-acetylcysteine. In the test tube, this agent is quite effective in dissolving mucin components and in decreasing the viscosity of sputum from patients with CF.80–82 Although some centers who have outstanding pulmonary outcomes, have used this agent as a standard part of the chronic regimen as an adjunct to airway clearance therapy in CF, others have been reluctant to use it because of a lack of randomized controlled trials.83 One CF center which has a track record of outstanding CF outcomes recommended using N-acetylcysteine in combination with sodium cromolyn and albuterol (W. Warwick, personal communication). We continue to recommend its use to our CF patients. Interestingly, N-acetylcysteine was found to activate CFTR Cl– conductance in cultured epithelial cells.84
In 1994, Pulmozyme, a DNA-cleaving enzyme, was approved for use in patients with CF following a large phase III multicenter trial.85 More than 900 patients were enrolled for a 6-month period. Three dosing regimens were employed: Placebo, 2.5 mg inhaled once daily, and 2.5 mg inhaled twice daily. The treatment groups showed a 5% improvement in FEV1 over placebo, as well as a slightly lower relative risk of exacerbation of lower respiratory tract infection after 6 months. There was no difference between the once- and twice-daily treatment groups. A second study revealed that Pulmozyme, inhaled once daily over 96 weeks, maintained pulmonary function and decreased the relative risk of respiratory tract exacerbations in young CF patients with normal FEV1 (≥85%).86 Currently, this drug is in fairly widespread use for CF. However, questions regarding patient selection and timing and duration of use of this expensive drug remain unanswered.
Abnormal homeostasis of airway surface fluid results in dehydrated secretions and impaired mucociliary clearance. As a strategy to improve airway surface hydration and airway clearance, inhaled hypertonic therapy was evaluated. Patients with CF, age 6 years and older, inhaled 7% hypertonic saline twice daily following a bronchodilator for 48 weeks; results revealed only a modest improvement in FEV1, but a significant reduction in the number of pulmonary exacerbations and days lost from school or work.87 Inhaled 7% hypertonic saline did not decrease the frequency of pulmonary exacerbations in CF infants and children less than 6 years of age,88 and therefore did not meet the primary outcome for approval as a maintenance therapy in this age group. However, in a subset of infants tested, infant pulmonary function testing revealed an improvement in FEV0.5 in the hypertonic saline group suggesting that this therapy may be useful and considered on an individual basis.
Bronchodilators and Anti-Inflammatory Agents
Bronchodilators are often used in treating the pulmonary manifestations of CF. Their use should be individualized. For example, in many patients, bronchospasm that is reversible with bronchodilators at one point in the course of the illness may prove refractory a short time later. Some patients undergo deterioration in pulmonary function following use of bronchodilators. In infants who are audibly wheezing, a bronchodilator can be tried. In older patients, pulmonary function testing provides a more objective and quantitative measure of bronchodilator effectiveness.
Corticosteroids have been used with good results in infants with severe obstructive airway disease that does not respond to antibiotics and bronchodilators and in patients with CF in whom the pulmonary disease is complicated by severe asthma or allergic bronchopulmonary aspergillosis (ABPA). Preliminary observations initially suggested that patients with CF would benefit from long-term administration of alternate-day corticosteroids, based on the presumption that corticosteroids would decrease the airway inflammatory response. However, in a large, placebo-controlled, multicenter trial of alternate-day corticosteroids administered in two dosage regimens (1 mg/kg and 2 mg/kg), the development of many side effects precluded a general recommendation for long-term corticosteroid treatment in CF.89 Subgroup analysis led to the suggestion that patients with moderately severe obstructive airway disease and those with chronic Pseudomonas infection might benefit from treatment for periods of less than 1 year. Beneficial effects were sufficient to prompt further studies of anti-inflammatory agents in CF. A controlled 4-year trial of high doses of ibuprofen in 40 patients with CF showed improvement in the rate of decline of pulmonary function in children.90 Questions remain whether side effects that might accrue with continued therapy will justify the gains. In concert, these two studies suggest that future development of a lung-specific anti-inflammatory agent with fewer systemic side effects may offer a promising approach.
Antibiotics
Two major innovations using antibiotics have been implemented as part of the regimen of maintenance therapy for CF. First, inhaled therapies have been demonstrated to successfully eradicate initial Pseudomonas infection and postpone chronic colonization in three large prospective trials of patients with CF in North America and in Europe.91–93 Second, chronic inhaled and/or oral antibiotic therapies successfully decrease the progression of lung disease related to chronic Pseudomonas infection. In addition to these therapies, it is important to emphasize that person-to-person transmission of P. aeruginosa and other opportunistic microbes is another source for chronic colonization. In healthcare facilities for CF patients, contact isolation precautions are recommended.94
Chronic airway colonization/infection with P. aeruginosa promotes progression of lung disease. Hoiby et al. championed early treatment of P. aeruginosa–positive sputum cultures with inhaled colistin and oral ciprofloxacin, even in the absence of symptoms, as a modality to prevent chronic colonization.95 The EPIC trial91 tested four randomized regimens: Cycled therapy versus culture-driven therapy; and 28 days of inhaled tobramycin inhalation solution (TIS) in the presence or absence of 14 days of oral ciprofloxacin. Approximately 80% of patients remained free of P. aeruginosa for the duration of the study (18 months) with no difference concerning time to first pulmonary exacerbation attributed to regularly cycled therapy or the addition of ciprofloxacin. The ELITE study92 was an open label randomized study comparing 28 to 56 days of inhaled TIS which revealed approximately 90% eradication at the end of therapy with 66% to 69% of patients having Pseudomonas-free cultures at the end of the 27-month study. In addition, a trial comparing inhaled colistin and oral ciprofloxacin to inhaled TIS and oral ciprofloxacin showed no difference between regimens in eradication with approximately 62% to 65% of patients with Pseudomonas-free cultures at 6 months.93 Therefore, although the evidence from these three studies supports antibiotic eradication of new Pseudomonas infection, there is no consensus yet for a specific therapeutic regimen.
Another approach that has been advocated is suppression of chronic Pseudomonas colonization by alternating monthly cycles of inhaled antibiotics. Inhaled, preservative-free TIS, 300 mg twice daily for 28 days on and 28 days off, improved pulmonary function (FEV1 increased by 10%) at the end of the third treatment cycle (20 weeks) compared to placebo.96 Recently, inhaled aztreonam, 75 mg three times per day, was tested in an open label study of cycling monthly regimen for patients with chronic P. aeruginosa infection.97 Patients reported improved symptoms and pulmonary function during on-therapy months with sustained weight gain over the 18-month duration of the study. Currently, prospective clinical trials are evaluating the efficacy of continuous alternating inhaled antibiotics compared to on–off cycling inhaled antibiotics.
Another antibiotic which has been studied as a chronic therapy in CF is azithromycin. Azithromycin, 250 mg or 500 mg thrice weekly, was evaluated in CF patients colonized with P. aeruginosa. After 6 months of therapy, patients on azithromycin had a modest improvement in FEV1 (6.2%), increased weight gain, and decreased rates of pulmonary exacerbations.98 Although macrolide antibiotics have been reported to have anti-inflammatory properties,99 there is no direct evidence of azithromycin-induced anti-inflammatory activity in CF. One concern about inhaled tobramycin, inhaled aztreonam, and oral azithromycin as chronic therapies is the risk of bacterial resistance and selection for growth of MDRO. In addition, questions regarding selection of patients and timing and duration of treatment remain unanswered.
CFTR Potentiators and Correctors
The most exciting breakthrough in CF therapies was announced in 2011 with the proof-of-concept demonstration that an oral drug, ivacaftor®, corrected the physiologic impact of a CFTR mutation, G551D. The G551D mutant CFTR protein is expressed at the cell surface but does not conduct chloride or regulate other ion channels. Ivacaftor® was discovered using a high throughput screening approach.100 In a phase III randomized, placebo-controlled, double-blind trial for CF patients with at least one copy of G551D mutation, ivacaftor®, administered orally, 150 mg twice per day for 48 weeks, increased FEV1 percent predicted by 10.6%, decreased risk of pulmonary exacerbations by 55%, improved CF quality of life (CFQL) respiratory symptom scores by 8.6 points, decreased sweat chloride values by 48 mmol/L, and was associated with an average 2.7-kg weight gain.67 Currently, other mutations similar to G551D are being tested as potential ivacaftor® targets. Importantly, other compounds that potentially correct F508del CFTR are being tested. One lead compound, VX-809 has been reported to decrease sweat chloride values in a dose-dependent manner.101 VX-809 will be combined with ivacaftor® for complementary combined therapy to enhance both processing and functioning of F508del-CFTR in homozygous patients.
Management of Acute Exacerbations of CF Bronchitis
During the past few decades of treatment of CF, antibiotics have proven to be the key element responsible for increased survival. A reasonable approach balances the dangers of overzealous administration of antibiotics against progressive airway damage and bronchiectasis resulting from untreated infection. The approach is based on sputum culture at the time of diagnosis and at regular intervals thereafter.
When signs and symptoms herald an exacerbation of pulmonary infection (i.e., increased cough or sputum production, dyspnea, decreased exercise tolerance, decreased appetite) or new abnormalities on the physical examination (i.e., increased respiratory rate, use of accessory muscles, changes on auscultation of the chest including decreased breath sounds, new crackles or wheezes, weight loss), new abnormalities on the chest radiograph, or a decline in pulmonary function tests, chest physiotherapy is increased and appropriate antibiotics are given orally, or for severe exacerbations, intravenously.
Currently useful agents for treating staphylococcal infections include dicloxacillin, cephalexin, the third-generation penicillin–clavulanic acid combinations, and macrolides. Early in the course of the pulmonary disease, a small fraction of Pseudomonas strains may be sensitive to tetracycline, trimethoprim/sulfamethoxazole, or chloramphenicol. Occasionally, even Pseudomonas strains considered resistant according to laboratory sensitivity tests apparently respond to these antibiotics. A mechanism that has been proposed to account for this phenomenon is that even though the antibiotic is not bactericidal, it may inhibit either growth of the organism or its production of exotoxin and proteases. Ciprofloxacin, a quinolone derivative that can be given orally, is initially effective against many strains of Pseudomonas and has gained widespread use in the outpatient management of CF. A major disadvantage in its use is that resistance often develops after a few courses of treatment.
For treatment of a severe pulmonary exacerbation of CF caused by methicillin-resistant Staphylococcus, vancomycin or linezolid are indicated. For Pseudomonas, a combination of an aminoglycoside given intravenously and a semisynthetic penicillin is generally used. This combination is presumed to act synergistically on Pseudomonas, and the Pseudomonas is less likely to become resistant to either antibiotic.
The most popular antibiotic combination currently in use is tobramycin and ceftazidime. To achieve high levels of antibiotics in the airways and in secretions, the aminoglycoside is generally administered in higher doses, 10 mg/kg/d instead of 7.5 mg/kg/d. A recent randomized trial comparing once-daily versus three-times–daily regimens of tobramycin revealed that once-daily IV therapy provided equivalent efficacy with less nephrotoxicity in children.102 Dosing should be titrated for serum peak levels of 20 to 30 mg/L and trough levels of 1 mg/L or less.
Third-generation penicillins and cephalosporins, piperacillin, and ceftazidime; carbapenems, imipenem and meropenem; and the latest β-lactam, aztreonam, are also quite effective against Pseudomonas. When given alone, resistance often develops quickly. Usually, these agents are used in combination with an aminoglycoside. Because the sensitivity and resistance patterns of the Pseudomonas often change, various combinations are tried at different times, with clinicians relying on sensitivities from recent isolates to determine which is most effective for the particular strain of Pseudomonas. For other resistant gram-negative organisms, such as B. cepacia, S. maltophilia, and Achromobacter xylosoxidans, other antibiotic combinations are indicated, including ceftazidime, meropenem, ciprofloxacin, minocycline, aztreonam, chloramphenicol, or trimethoprim/sulfamethoxazole.
Staphylococcus, Pseudomonas, and other gram-negative organisms, such as B. cepacia, A. xylosoxidans, and S. maltophilia, once found in the sputum, are rarely eradicated. However, most other manifestations of an exacerbation of pulmonary disease abate during a 2-week course of antibiotics administered intravenously; for example, the densities seen on the chest radiograph decrease, the white blood cell count decreases, fever and respiratory rate decrease, and pulmonary function test results, which often deteriorate at the start of an exacerbation, return to their previous baseline. Although many patients begin to show improvement after 5 to 7 days, most CF centers continue antibiotics intravenously for at least 2 weeks to decrease the relapse rate and to avoid a decrease in the interval between exacerbations. Indeed, some centers routinely recommend a 3- to 4-week course of intravenous antibiotics to treat an exacerbation of a pulmonary infection. In the occasional-hospitalized patient who experiences a relapse or manifests an increase in symptoms shortly after administration of intravenous antibiotics is stopped, long-term intravenous administration of an aminoglycoside can be continued with use of a heparin lock. This technique may be helpful in allowing the patient to return home while still receiving effective doses of aminoglycosides.
Nutritional Support
Patients with CF should have a detailed nutrition assessment at diagnosis and annually as per the CFF guidelines.60 Nutritional status should be screened at every visit. Patients are prescribed a high-calorie balanced diet. The CFF recommends that for infants and young children (0–2 years) weight for length be maintained at the 50th percentile and that children and adolescents (2–20 years of age) have their BMI percentile at the 50th percentile. Although it is true that pulmonary function is the predominant factor in determining morbidity and mortality in CF, it is becoming increasingly clear that overall patient status is closely tied to nutritional status. Importantly, achieving and maintaining normal nutritional status is associated with maintenance of lung function in young children and adults. Calorie goals are often 110% to 120% of usual calorie requirements. At these caloric intakes, protein intake is often adequate to meet needs. Patients are encouraged to achieve calorie goals by the ingestion of calorically dense foods. If this is hard to achieve, calorie boosters (vegetable oils, butter, and cheese) are recommended followed by the use of high-calorie supplements (shakes). Calorie needs may be increased in patients with chronic lung disease, malabsorption, and chronic liver disease. Nocturnal nasogastric feeds may be used in the short term for aggressive nutritional rehabilitation, and patients who need long-term support have placement of a gastrostomy tube for ease of care. Typically standard formulas are used but some patients with feeding intolerance and poor weight gain benefit from hydrolyzed formulas. Intravenous or parenteral nutrition is rarely used, but may be required in patients who have had GI surgery.
Pancreatic status (insufficiency or sufficiency) should be determined and monitored as needed. The mainstay in managing the pancreatic insufficiency of CF is PERT which consists of enteric-coated capsules containing amylases, proteases, and lipases. PERT should be ingested before meals that contain protein, fat, or complex carbohydrates. Dosing guidelines have been developed by the CFF. Fibrosing colonopathy or the development of colonic strictures is a complication that appears to be related to high PERT doses exceeding 10,000 lipase units/kg/d and was first noted following the introduction of high-potency pancreatic enzymes.103 Most patients can be managed with pancreatic enzyme doses within the published guidelines. Patients who require higher pancreatic enzyme doses should be evaluated by a dietitian and a pediatric gastroenterologist.
Patients with pancreatic insufficiency are at risk for fat-soluble vitamin deficiency. Guidelines also exist for vitamin supplementation and CF-specific vitamin products are available.60 It is recommended that fat-soluble vitamin status be monitored annually and additional supplements (Vitamin D) may be required if levels are still low. It is very unusual for patients with CF to be vitamin A deficient if they are on a CF-specific vitamin preparation. Supplemental salt is needed by patients to prevent salt depletion. Salt is added to infant formula and children and adults are encouraged to salt their foods liberally and to take salt-containing liquids and snacks during hot weather and periods of increased physical activity.
 MANAGEMENT OF CYSTIC FIBROSIS–RELATED DIABETES
MANAGEMENT OF CYSTIC FIBROSIS–RELATED DIABETES
Insulin is the treatment of choice for CFRD, and ideally the regimen is customized to fit the needs of the individual patient. Combinations of basal (long-acting) and bolus (rapid-acting) insulins are used in the treatment of CFRD with FH. In the absence of FH, premeal rapid-acting insulin is the main treatment approach. Frequent meals, snacking, and “grazing” are not uncommon in CF, and the requirement of multiple daily injections can be prohibitive for some individuals. The insulin pump offers flexibility and can negate the need for frequent injections.104 Moreover, calories are not restricted in CFRD although either avoidance of foods of low nutritional value (sugared soda or confection) or their consumption in combination with complex carbohydrates, protein, and fats is useful in avoiding excessive hyperglycemia. Pancreatic enzyme replacement also appears to improve meal-related glucose excursion.36 The role of treatment of prediabetes and early insulin deficiency in preserving pancreatic β-cell function, pulmonary function, and nutritional status has yet to be defined.
NATURAL HISTORY AND PROGNOSIS
A comprehensive treatment program for CF has unequivocally improved overall survival of patients. Fifty years ago, the median survival was only a few years of age. For the 5-year period from 2007 to 2011 the median predicted survival was 36.8 years of age (CF Foundation Patient Registry, 2011 Annual Data Report, Bethesda, MD). However, because CF is a complex disorder that affects different organs to different degrees, it is difficult to describe a “typical course” for a patient with CF. Some patients die in childhood or adolescence, while others survive well beyond age 40.
An important determinant of the natural history of CF is the severity of the pulmonary disease and the rate at which it progresses. Although most patients’ condition improves in response to therapy, skillful management does less to influence the course of the severely affected than that of the mildly affected patient.
A variety of scoring systems have been devised for CF. The clinical scoring system devised by Shwachman and Kulczycki and the chest radiograph scoring system devised by Brasfield and associates are widely used. However, although these and more elaborate scoring systems are useful in categorizing patients according to the severity of illness, none has proved useful in prognosticating the course of an individual patient.
Because CF is a genetic disease, the question of a familial pattern of severity is often raised. Figure 50-7 shows chest radiographs of three siblings with CF; the radiographs demonstrate mild, moderate, and severe disease in individuals in the same family. The capsule histories, which are included in the figure legend, also illustrate the variability in courses experienced.
Patients with CF can be categorized not only with respect to severity of illness, but also with regard to survival. For example, more than half of patients with CF who underwent surgery for meconium ileus before 1965 died in the first 2 months of life. Although this situation had improved markedly by 1976, the survival rate for patients with meconium ileus was still not as good as for all other patients with CF. In addition, the survival rate was much lower for females than for males, especially in adolescents. In recent years, differences between the patients in these groups have declined or disappeared. Because of improvements in the collection of mortality statistics, comparison of current data with those from previous years may be somewhat misleading, but 50% survival age has not been increasing as rapidly in recent years as in the 1970s and 1980s. Furthermore, there is a difference in outcomes among individual CF centers.
COMPLICATIONS
The course of CF is often characterized by a gradual decrease in pulmonary function, punctuated by further abrupt declines during exacerbations. Malnutrition, when present despite therapy, usually correlates best with the severity of the pulmonary disease. However, the course of CF may be suddenly altered by certain complications of the disease.
 HYPOELECTROLYTEMIA AND METABOLIC ALKALOSIS
HYPOELECTROLYTEMIA AND METABOLIC ALKALOSIS
Hypoelectrolytemia and metabolic alkalosis are serious complications that are especially apt to occur during periods of hot weather, when losses of sodium and chloride increase. Electrolyte depletion may be life-threatening, especially in infants and young children (Table 50-3). Prompt fluid replacement with isotonic saline is critical.



