•Adverse local tissue interactions:
– Inflammation
– Toxicity
– Carcinogenic response
– Calcification
– Embolization or lymphatic spread of material fragments
•Induced device migrations: encapsulation or foreign body response
•Inappropriate or altered healing responses
•Associated infections
•Thrombosis
– Thrombotic occlusion
– Thromboembolism
The mechanisms related to how the host or patient may respond to different components of a cardiac device must be extensively evaluated to ensure that appropriate materials are selected for the final device design. This evaluation can be complicated by the fact that it is hard to replicate the range of human immune responses that may exist in vitro. To address this, all cardiac devices must also undergo rigorous animal testing, although it is important to note that animal testing can sometimes provide misleading results on species-specific bioreactivity. An example of this can be seen in the design and development of the Braunwald–Cutter heart valve ball and cage prostheses (Fig. 42.1), whereby cloth-covered cage struts were designed to encourage endothelialization and subsequently decrease any chance of thrombolytic events [3]. Extensive testing in the mitral position of pigs, sheep, and calves showed promising results; thus, the device was approved for human clinical trials. However, the device did not elicit the same host responses when implanted in humans; rather, it resulted in aggravated wear on the cloth cage struts and, more critically, the formation of debris embolization.

Fig. 42.1
Braunwald–Cutter valve
It should also be emphasized that host responses are not limited to immune reactions. For example, implanted tissue heart valves remain susceptible to accelerated prosthesis leaflet calcification. More specifically, it has been reported that this type of calcification is initiated by reactions between the extracellular fluid and the leaflet membranes, creating calcium phosphate mineral deposits [4]. As a result, much research is ongoing to enhance leaflet materials and minimize bioreactivity, including calcification inhibitors on the valves that limit mineral deposition on the implanted materials. Furthermore, the materials used in a device can display unexpected interactions with the host, as evidenced by the earliest versions of the Starr–Edwards caged-ball mechanical heart valve. The valve design used a silicone ball that was found to absorb lipids from the blood and thus swell [4]. In addition to resultant poor valve function, this also caused the silicone balls to become brittle; in turn, this increased the possibility of ball fracture and consequent embolization of small fragments into the arteries downstream of the valve position.
42.2.3 Durability
Along with the biocompatibility, a device must be durable enough to withstand the mechanical loads placed upon it. These forces can be exerted upon the device by implantation, cardiac motion, or external events such as getting hit in the chest or falling on the ground (specifically for implantable devices). Potential situations like these must be considered when designing the device, to ensure it has the robustness needed to withstand such environments as well as an active lifestyle.
Major factors that any design engineer must take into consideration are the repetitive movements and strains that will be placed on a device within a patient’s body. If you were to imagine the environment in which a coronary stent is placed, the arteries will have a cyclic pressure placed on them which, in turn, will flex and relax the stent with each heartbeat. This requires the stent’s material to either: (1) oscillate with the artery while being durable enough to withstand flexing for the lifetime of the stent or (2) provide enough opposing force to the artery that it does not move during these times of increased pressure.
Cardiac pacing and defibrillation leads are also exposed to repetitive mechanical stresses that can cause faults or failures in their integrity. Two individual incidents were observed within defibrillation leads from two separate companies—St. Jude Medical (St. Paul, MN, USA) and Medtronic, Inc. (Minneapolis, MN, USA)—that were competing to get a small defibrillation lead on the market. The two companies produced small-diameter leads, the Riata (St. Jude Medical) and Sprint Fidelis (Medtronic, Inc.), that gained FDA approval and were then appropriately implanted clinically [5]. Before the release of these smaller leads, >8F defibrillation leads were the standard of care, but in 2002, St. Jude Medical released their 7.6F Riata defibrillation lead. In response, Medtronic, Inc., released their 6.7F Sprint Fidelis in 2004, shortly followed by the 6.3F Riata ST by St. Jude Medical. The perception was that a smaller lead would decrease the blockage caused within the venous system at the level of the subclavian vein. However, it was found that these systems were unable to perform at adequate levels of durability within the body [5]. Both companies ended up having issues where their leads broke or eroded to the point where the conductor wires could penetrate and cause misreading of the cardiac signals and potentially lead to excessive shocks being delivered to the patient (or even worse, no shocks were delivered when they were needed).
In 2007, Medtronic, Inc., sent a letter to physicians informing them that they had seen failures in the field both at the distal portions of these leads and at points near the anchoring sleeve tie-down. Further, they reported that the distal failures were significantly affected by the bending of the lead body due to tortuous anatomy in the veins. The failures at the tie-down location were potentially caused by the way physicians implanted and secured the leads within the body. Each of these failure modes suggested that the way in which the lead was implanted could make a significant impact on the longevity of these devices and their potential for failure. Ultimately in late 2007, Medtronic, Inc. issued a voluntary market suspension that the FDA formally considered to be a recall. During this time, St. Jude Medical was also seeing similar issues with their leads. They reported that portions of their leads were being eroded to the point where the wires were being externalized in both styles of the Riata leads. It was also reported that the primary reason for this externalization was due to abrasion of the lead insulation with either the anatomy (e.g., tricuspid valve) or the internal components of the lead (e.g., the pacing/sensing wires). In one study, they found that a common defect was due to the internal abrasion of either the pace/sense coils or the shocking coils against the silicone insulation. This would be in response to the repetitive motion of the lead and the ability for the internal components to move relative to the external insulation. This caused what they deemed to be an inside-out abrasion, where these wires were wearing away at the insulation and become externalized and readily visible under fluoroscopy [6]. Due to these failures, St. Jude Medical ended up removing the Riata family of leads from the market in 2010 and recalling them in 2011. Since these recalls, the defibrillator lead market has stayed at >8F, due to the perception that smaller leads could be associated with potential failures in patients.
Both of these cases are prime examples for why device durability testing is essential for the development of robust cardiac devices. Notice that it is not always a matter of how the device will react within the body (which seemed to be the case for the Riata leads), but also how the devices are implanted that may lead to potential failure modes. Ultimately, if a cardiac device is not tested properly in relevant use conditions, certain factors can be overlooked and may ultimately end up negatively impacting patients.
42.2.4 Design for Manufacture
The ability to put together a device, or manufacturability, is something that should not be overlooked during the design processes. By incorporating early feedback about manufacturing the product, the risks of not being able to build the device efficiently enough or to produce adequate quantities to fulfill demand will be greatly reduced. Failure in either case could cause the product to be stifled, no matter how successful or clinically helpful it is. As such, design for manufacturability (DFM) has become a common practice in the cardiac device industry, a practice that emphasizes how a successful design should ensure the highest-quality products while decreasing manufacturing costs. This is accomplished by making manufacturing cost estimates to proactively guide and prioritize cost reduction efforts involved with design. Consequently, DFM should have significant effects on product lead times, development costs, and ultimate product quality. As such, DFM specifically requires input from a multidisciplinary team, including manufacturing engineers, cost accountants, and production personnel, in addition to the design engineers [7]. Yet, when applying these principles to cardiac device design, it is imperative to understand that the quality of care impacted by the device must not be compromised by the need for a more cost-effective manufacturing process. One needs to consider that production costs can be controlled by using existing technologies and established manufacturing techniques. There are many methods for ensuring that the requirements for manufacturing are being taken into consideration, and there are several numerical methods of design and testing (also known as in silico testing) that can be used to streamline the design for the manufacturing process.
42.3 Development Process
There are often two types of development referred to by designers—iterative and disruptive. Those that fall into the iterative category are devices that generally are perceived as logical next steps in the development chain. For example, this would include the addition of a third and fourth electrode to a left-sided lead that would give the physician more vectors to choose from when programming a pacemaker, as discussed in Chap. 30. The ideas and devices that are considered to be disruptive are those that medical industry professionals would consider as game changers . These are devices like transcatheter-delivered cardiac valves. The option prior to these transcatheter valves was open-heart surgery, which often involved placing the patient on cardiac bypass and fully stopping the heart; this procedure limited the population of patients who could receive the treatment to those that were able to physically undergo an invasive surgery and required weeklong recovery periods in the hospital. In other words, these devices have effectively provided a means to treat more of the patient populations which were underserved by prior medical management.
Although there may be a valid unmet clinical need, oftentimes there are points along the product development pathway that may ultimately put a halt to a device or therapy. This can be as simple as the product not working as intended or a business decision to not pursue a particular therapy because it does not align with their internal objectives. This, along with other reasons, is generally why larger companies will often focus their efforts on incremental improvement of devices as opposed to disruptive technologies. The incremental improvements are much more predictable and lower risk, since the market is already known and the needs often come directly from their voice of customer (VOC) . Disruptive technology offers a greater amount of risk and, conversely, often a higher reward. This volatility of the device can be a deterrent for larger, publicly held companies that rely on stability, but it provides an opportunity for smaller companies willing to take the risk.
Whether it is an iterative or disruptive technology, the development process generally follows a similar pathway (also utilized in nonmedical device development), with the hope of producing a marketable product. Often, a problem is identified within the field, and to better understand the clinical needs, the designers must empathize with those experiencing the problem. This can be the physician, patient, payers, or any other stakeholders. While studying and understanding these situations, the designers gain invaluable insights as to the root cause(s) of the clinical problem or need, as well as what confounding factors are present. Then, by defining which portion of the issue they are trying to address, designers can begin to hone in on a potential solution. In turn, this will spark ideas and a multitude of solutions (partial or complete) will be generated. These solutions (as you will see in Sect. 42.3.3) will be prototyped and selected based on technical and market feasibility. Promising options will then be tested for usability and functionality; if an acceptable option is produced, the design will be frozen to move on to preclinical animal testing and eventual clinical testing (Fig. 42.2) (see Chap. 43). The generalized overarching progression of how cardiac devices develop from concept to market will be presented in the following sections.
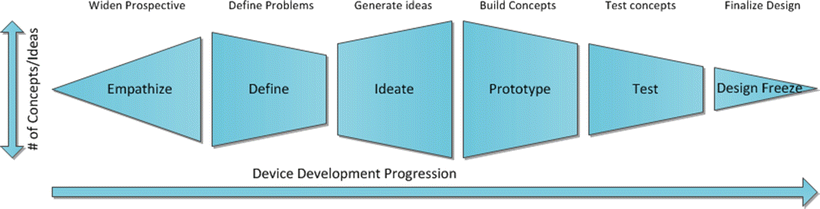
Fig. 42.2
Flowchart of the design thinking process
42.3.1 Six-Phase Approach to Device Development
There are many ways that device development can be approached, but often they follow a similar pattern: (1) generate an idea, (2) prove it to be feasible, (3) test the idea, (4) market the idea, (5) and make sure that it does not cause unfavorable issues in the field. This is obviously oversimplifying things, and each of these steps requires a vast amount of work to “get it right.” Despite the simplification, all of these steps are essential to fully develop and market a cardiac device. This section will describe a more commonly used phase–gate system that breaks up the process into six phases of development, as shown in Fig. 42.3. Importantly, throughout each of these phases, a number of different elements need to be checked and rechecked in order to create a successful device.
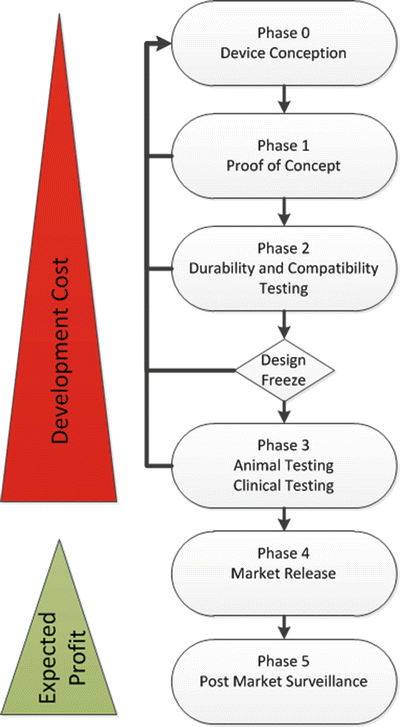
Fig. 42.3
Flowchart of the device development process . The six phases of device design are shown, as well as the associated cost and profit factors related to marketing a medical device
As the development process advances along the phases, the subsequent tasks are generally associated with higher costs. For instance, the first phase of the development process is device conception , in which a clay model or a sketch of the product may be the only thing required, along with asking some key stakeholders whether or not the idea is worth the time and effort. This requires very little monetary investment. The next phases require creating animations and prototypes and testing of these advanced concept devices; these phases will incur greater costs. Prior to market release, at a minimum, preclinical and even some clinical testing will be necessary, which may require the investment of millions of dollars and multiple years of study before finally being able to market and realize profit from the device.
Phase 0 is the planning phase during which much of the groundwork is completed, including developing prototype devices, creating a product platform, assessing market opportunities (determining if it will be worth pursuing financially), and identifying product constraints associated with intellectual property (see Sect. 42.3.2). The decision to move on to the next phase often requires insights related to perceived market value versus return on investment, an evaluation that is required to bring any device to the market.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


